
Genetic Determinants of Acinetobacter baumannii Serum-Associated Adaptive Efflux-Mediated Antibiotic Resistance
 , , , and
, , , and
Abstract
:1. Importance
2. Introduction
3. Materials and Methods
3.1. Bacterial Strains and Growth Conditions
3.2. Transposon Mutant Screening for Loss of Adaptive Efflux-Associated Resistance
3.3. Identification of Transposon Insertion Site of Mutants of Interest
3.4. Adaptive Antibiotic Tolerance Assays
3.5. Ethidium Bromide Efflux Assays
3.6. Outer Membrane Integrity Assay
3.7. Construction of Complementation Plasmids
3.8. Real-Time Quantitative PCR to Measure A1S_3277 Expression
3.9. Synthesis of DMACA-Ofloxacin
3.10. Flow Cytometry
3.11. Confocal Microscopy
3.12. RNA Sequencing
3.13. Statistics
4. Results and Discussion
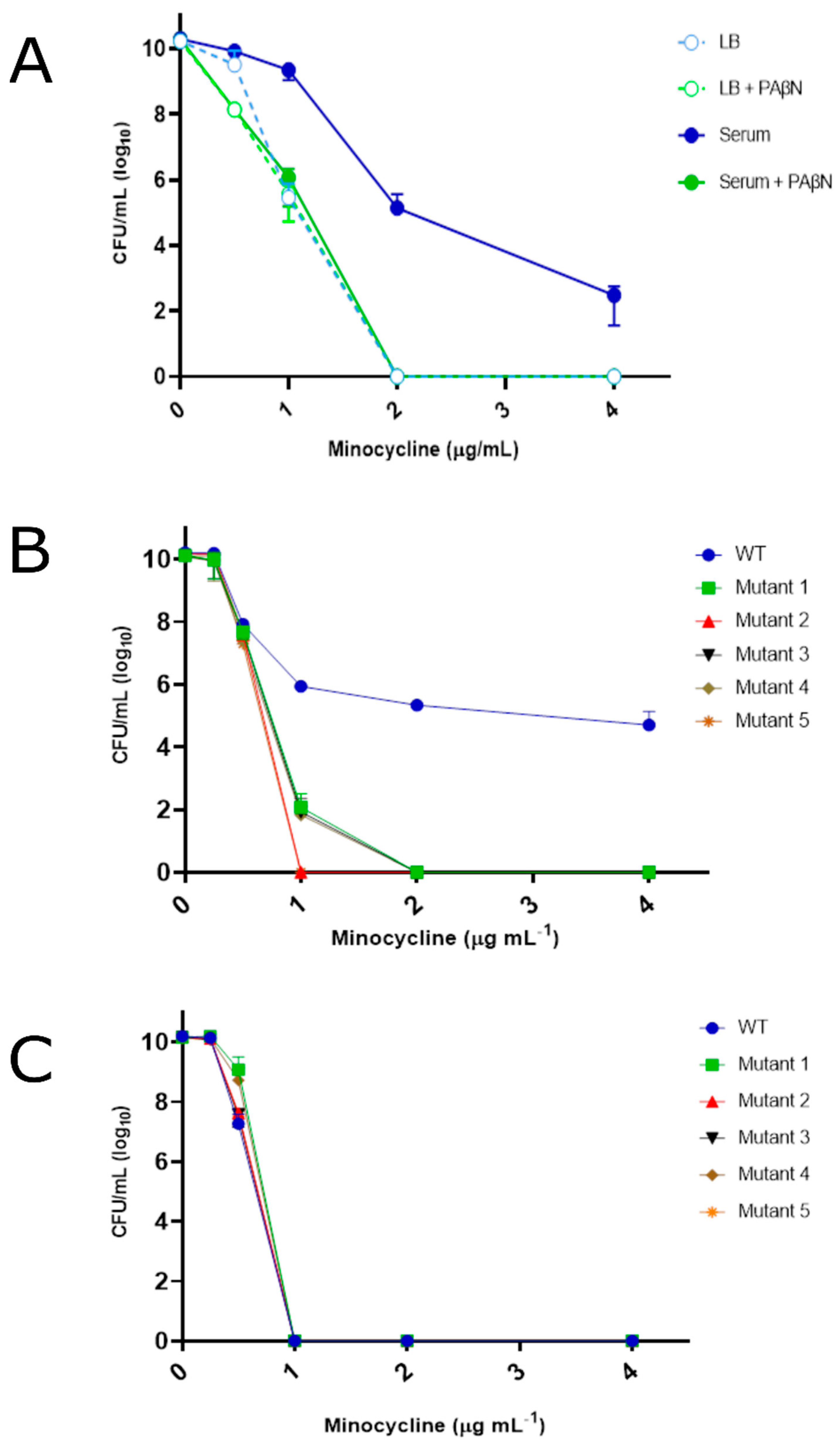
4.1. YhaK Impacts A. baumannii Serum-Inducible Efflux Properties
4.2. YhaK Impacts A. baumannii Serum-Inducible Adaptive Efflux-Mediated Antibiotic Resistance
4.3. YhaK-Mutant Cells Accumulate DMACA-Oflox
4.4. RNA Sequencing of Wild-Type and YhaK-Mutant Cells during Growth in Human Serum
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Verma, P.; Tiwari, M.; Tiwari, V. Efflux pumps in multidrug-resistant Acinetobacter baumannii: Current status and challenges in the discovery of efflux pumps inhibitors. Microb. Pathog. 2021, 152, 104766. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Breidenstein, E.B.; Hancock, R.E. Creeping baselines and adaptive resistance to antibiotics. Drug Resist. Updat. 2011, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hornsey, M.; Ellington, M.J.; Doumith, M.; Thomas, C.P.; Gordon, N.C.; Wareham, D.W.; Quinn, J.; Lolans, K.; Livermore, D.M.; Woodford, N. AdeABC-mediated efflux and tigecycline MICs for epidemic clones of Acinetobacter baumannii. J. Antimicrob. Chemother. 2010, 65, 1589–1593. [Google Scholar] [CrossRef] [Green Version]
- Metan, G.; Alp, E.; Yildiz, O.; Percin, D.; Aygen, B.; Sumerkan, B. Clinical experience with tigecycline in the treatment of carbapenem-resistant Acinetobacter infections. J. Chemother. 2010, 22, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; Potoski, B.A.; Rea, R.; Adams, J.; Sethi, J.; Capitano, B.; Husain, S.; Kwak, E.J.; Bhat, S.V.; Paterson, D.L. Acinetobacter baumannii bloodstream infection while receiving tigecycline: A cautionary report. J. Antimicrob. Chemother. 2007, 59, 128–131. [Google Scholar] [CrossRef]
- Rice, L.B. Challenges in identifying new antimicrobial agents effective for treating infections with Acinetobacter baumannii and Pseudomonas aeruginosa. Clin. Infect. Dis. 2006, 43 (Suppl. S2), S100–S105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, M.I.; Jacobs, A.C.; Sayood, K.; Dunman, P.M.; Skaar, E.P. Acinetobacter baumannii increases tolerance to antibiotics in response to monovalent cations. Antimicrob. Agents Chemother. 2010, 54, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.C.; Sayood, K.; Olmsted, S.B.; Blanchard, C.E.; Hinrichs, S.; Russell, D.; Dunman, P.M. Characterization of the Acinetobacter baumannii growth phase-dependent and serum responsive transcriptomes. FEMS Immunol. Med. Microbiol. 2012, 64, 403–412. [Google Scholar] [CrossRef] [Green Version]
- CDC. Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA, 2019; pp. 1–150.
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Wellcome Trust and HM Government: London, UK, 2016.
- Hidron, A.I.; Edwards, J.R.; Patel, J.; Horan, T.C.; Sievert, D.M.; Pollock, D.A.; Fridkin, S.K. NHSN annual update: Antimicrobial-resistant pathogens associated with healthcare-associated infections: Annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 2008, 29, 996–1011. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Ye, G.; Olesky, M.; Lawrence, K.; Murray, J.; Yu, K. Trends in resistant Enterobacteriaceae and Acinetobacter species in hospitalized patients in the United States: 2013–2017. BMC Infect. Dis. 2019, 19, 742. [Google Scholar] [CrossRef] [Green Version]
- Abadi, A.T.B.; Rizvanov, A.A.; Haertle, T.; Blatt, N.L. World Health Organization Report: Current Crisis of Antibiotic Resistance. BioNanoScience 2019, 9, 778–788. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Fournier, P.E.; Vallenet, D.; Barbe, V.; Audic, S.; Ogata, H.; Poirel, L.; Richet, H.; Robert, C.; Mangenot, S.; Abergel, C.; et al. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet. 2006, 2, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, E.J.; Balloy, V.; Fiette, L.; Chignard, M.; Courvalin, P.; Grillot-Courvalin, C. Contribution of the Ade Resistance-Nodulation-Cell Division-Type Efflux Pumps to Fitness and Pathogenesis of Acinetobacter baumannii. mBio 2016, 7, e00697-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, K.; Yamaguchi, A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 2001, 183, 5803–5812. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, I.T.; Sliwinski, M.K.; Saier, M.H., Jr. Microbial genome analyses: Global comparisons of transport capabilities based on phylogenies, bioenergetics and substrate specificities. J. Mol. Biol. 1998, 277, 573–592. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, M.; Chan, E.W.C.; Chen, S. Membrane Transporters of the Major Facilitator Superfamily Are Essential for Long-Term Maintenance of Phenotypic Tolerance to Multiple Antibiotics in E. coli. Microbiol. Spectr. 2021, 9, e0184621. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Horiyama, T.; Zhang, Y.; Li, X.; Nishino, K.; Yan, A. The Multidrug Efflux Pump MdtEF Protects against Nitrosative Damage during the Anaerobic Respiration in Escherichia coli. J. Biol. Chem. 2011, 286, 26576–26584. [Google Scholar] [CrossRef] [Green Version]
- Adams, K.N.; Takaki, K.; Connolly, L.E.; Wiedenhoft, H.; Winglee, K.; Humbert, O.; Edelstein, P.H.; Cosma, C.L.; Ramakrishnan, L. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 2011, 145, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Kristoffersen, S.M.; Ravnum, S.; Tourasse, N.J.; Økstad, O.A.; Kolstø, A.B.; Davies, W. Low concentrations of bile salts induce stress responses and reduce motility in Bacillus cereus ATCC 14579. J. Bacteriol. 2007, 189, 5302–5313. [Google Scholar] [CrossRef] [Green Version]
- Urdaneta, V.; Casadesús, J. Adaptation of Salmonella enterica to bile: Essential role of AcrAB-mediated efflux. Environ. Microbiol. 2018, 20, 1405–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.; Hancock, R.E. Adaptive and mutational resistance: Role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.M.; Chojnacki, M.; Fadrowski, J.J.; Bauza, C.; Dunman, P.M.; Dudas, R.A.; Goldenberg, N.A.; Berman, D.M. Serum-Associated Antibiotic Tolerance in Pediatric Clinical Isolates of Pseudomonas aeruginosa. J. Pediatric Infect. Dis. Soc. 2020, 9, 671–679. [Google Scholar] [CrossRef]
- Blanchard, C.; Barnett, P.; Perlmutter, J.; Dunman, P.M. Identification of Acinetobacter baumannii serum-associated antibiotic efflux pump inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6360–6370. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.C.; Blanchard, C.E.; Catherman, S.C.; Dunman, P.M.; Murata, Y. An ribonuclease T2 family protein modulates Acinetobacter baumannii abiotic surface colonization. PLoS ONE 2014, 9, e85729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, A.C.; Hood, I.; Boyd, K.L.; Olson, P.D.; Morrison, J.M.; Carson, S.; Sayood, K.; Iwen, P.C.; Skaar, E.P.; Dunman, P.M. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 2010, 78, 1952–1962. [Google Scholar] [CrossRef] [Green Version]
- Hunger, M.; Schmucker, R.; Kishan, V.; Hillen, W. Analysis and nucleotide sequence of an origin of DNA replication in Acinetobacter calcoaceticus and its use for Escherichia coli shuttle plasmids. Gene 1990, 87, 45–51. [Google Scholar] [CrossRef]
- Gallagher, L.A.; Ramage, E.; Weiss, E.J.; Radey, M.; Hayden, H.S.; Held, K.G.; Huse, H.K.; Zurawski, D.V.; Brittnacher, M.J.; Manoil, C. Resources for Genetic and Genomic Analysis of Emerging Pathogen Acinetobacter baumannii. J. Bacteriol. 2015, 197, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Helander, I.M.; Mattila-Sandholm, T. Fluorometric assessment of gram-negative bacterial permeabilization. J. Appl. Microbiol. 2000, 88, 213–219. [Google Scholar] [CrossRef]
- Chojnacki, M.; Cao, X.; Young, M.; Fritz, R.N.; Dunman, P.M.; Flaherty, D.P. Optimization of 4-Substituted Benzenesulfonamide Scaffold To Reverse Acinetobacter baumannii Serum-Adaptive Efflux Associated Antibiotic Tolerance. ChemMedChem 2020, 15, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Perdigão Neto, L.V.; Oliveira, M.S.; Orsi, T.D.; Prado, G.; Martins, R.C.R.; Leite, G.C.; Marchi, A.P.; Lira, E.S.; Côrtes, M.F.; Espinoza, E.P.S.; et al. Alternative drugs against multiresistant Gram-negative bacteria. J. Glob. Antimicrob. Resist. 2020, 23, 33–37. [Google Scholar] [CrossRef]
- Beganovic, M.; Daffinee, K.E.; Luther, M.K.; LaPlante, K.L. Minocycline Alone and in Combination with Polymyxin B, Meropenem, and Sulbactam against Carbapenem-Susceptible and -Resistant Acinetobacter baumannii in an In Vitro Pharmacodynamic Model. Antimicrob. Agents Chemother. 2021, 65, e01680-20. [Google Scholar] [CrossRef]
- Singh, S.; Khanna, D.; Kalra, S. Minocycline and Doxycycline: More Than Antibiotics. Curr. Mol. Pharmacol. 2021, 14, 1046–1065. [Google Scholar] [CrossRef] [PubMed]
- Seok, H.; Choi, W.S.; Lee, S.; Moon, C.; Park, D.W.; Song, J.Y.; Cheong, H.J.; Kim, J.; Kim, J.Y.; Park, M.N.; et al. What is the optimal antibiotic treatment strategy for carbapenem-resistant Acinetobacter baumannii (CRAB)? A multicentre study in Korea. J. Glob. Antimicrob. Resist. 2021, 24, 429–439. [Google Scholar] [CrossRef]
- Gilani, M.; Latif, M.; Gilani, M.; Saad, N.; Ansari, M.; Gilani, M.; Waseem, H.; Naeem, A. Efficacy of Antimicrobials Against Multidrug-Resistant Acinetobacter baumannii from Patients in a Tertiary Care Hospital. Microb. Drug Resist. 2020, 26, 681–684. [Google Scholar] [CrossRef]
- Ku, N.S.; Lee, S.H.; Lim, Y.S.; Choi, H.; Ahn, J.Y.; Jeong, S.J.; Shin, S.J.; Choi, J.Y.; Choi, Y.H.; Yeom, J.S.; et al. In vivo efficacy of combination of colistin with fosfomycin or minocycline in a mouse model of multidrug-resistant Acinetobacter baumannii pneumonia. Sci. Rep. 2019, 9, 17127. [Google Scholar] [CrossRef] [Green Version]
- Flamm, R.K.; Shortridge, D.; Castanheira, M.; Sader, H.S.; Pfaller, M.A. In Vitro Activity of Minocycline against U.S. Isolates of Acinetobacter baumannii-Acinetobacter calcoaceticus Species Complex, Stenotrophomonas maltophilia, and Burkholderia cepacia Complex: Results from the SENTRY Antimicrobial Surveillance Program, 2014 to 2018. Antimicrob. Agents Chemother. 2019, 63, e01154-19. [Google Scholar] [CrossRef]
- Dillon, N.; Holland, M.; Tsunemoto, H.; Hancock, B.; Cornax, I.; Pogliano, J.; Sakoulas, G.; Nizet, V. Surprising synergy of dual translation inhibition vs. Acinetobacter baumannii and other multidrug-resistant bacterial pathogens. EBioMedicine 2019, 46, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Fragkou, P.C.; Poulakou, G.; Blizou, A.; Blizou, M.; Rapti, V.; Karageorgopoulos, D.E.; Koulenti, D.; Papadopoulos, A.; Matthaiou, D.K.; Tsiodras, S. The Role of Minocycline in the Treatment of Nosocomial Infections Caused by Multidrug, Extensively Drug and Pandrug Resistant Acinetobacter baumannii: A Systematic Review of Clinical Evidence. Microorganisms 2019, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, K.J.; Eady, E.A.; Cove, J.H.; Taylor, J.P.; Cunliffe, W.J. Comparison of serum antibiotic levels in acne patients receiving the standard or a modified release formulation of minocycline hydrochloride. Clin. Exp. Dermatol. 1997, 22, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Rehmani, I.; Esaki, S.; Fu, R.; Chen, L.; de Serrano, V.; Liu, A. Pirin is an iron-dependent redox regulator of NF-κB. Proc. Natl. Acad. Sci. USA 2013, 110, 9722–9727. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Nunnelly, D.A.; Muhammad, D.; Mezzich, R.; Lee, B.S.; Jayathilaka, L.; Kaufman, L.S.; Warpeha, K.M. Pirin1 (PRN1) is a multifunctional protein that regulates quercetin, and impacts specific light and UV responses in the seed-to-seedling transition of Arabidopsis thaliana. PLoS ONE 2014, 9, e93371. [Google Scholar] [CrossRef] [Green Version]
- Midgley, M. The phosphonium ion efflux system of Escherichia coli: Relationship to the ethidium efflux system and energetic studies. J. Gen. Microbiol. 1986, 132, 3187–3193. [Google Scholar] [CrossRef] [Green Version]
- Stone, M.R.L.; Masi, M.; Phetsang, W.; Pagès, J.M.; Cooper, M.A.; Blaskovich, M.A.T. Fluoroquinolone-derived fluorescent probes for studies of bacterial penetration and efflux. Medchemcomm 2019, 10, 901–906. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.J.; Huang, D.Q.; Bai, Y.H.; Shen, Y.Y.; Lin, X.Z.; Huang, Y.; Ling, Y.R.; Fan, N.S.; Jin, R.C. How anammox process resists the multi-antibiotic stress: Resistance gene accumulation and microbial community evolution. Sci. Total Environ. 2022, 807, 150784. [Google Scholar] [CrossRef]
- Yamada, J.; Yamasaki, S.; Hirakawa, H.; Hayashi-Nishino, M.; Yamaguchi, A.; Nishino, K. Impact of the RNA chaperone Hfq on multidrug resistance in Escherichia coli. J. Antimicrob. Chemother. 2010, 65, 853–858. [Google Scholar] [CrossRef]
- Deter, H.S.; Abualrahi, A.H.; Jadhav, P.; Schweer, E.K.; Ogle, C.T.; Butzin, N.C. Proteolytic Queues at ClpXP Increase Antibiotic Tolerance. ACS Synth. Biol. 2020, 9, 95–103. [Google Scholar] [CrossRef]
- Dechend, R.; Hirano, F.; Lehmann, K.; Heissmeyer, V.; Ansieau, S.; Wulczyn, F.G.; Scheidereit, C.; Leutz, A. The Bcl-3 oncoprotein acts as a bridging factor between NF-kappaB/Rel and nuclear co-regulators. Oncogene 1999, 18, 3316–3323. [Google Scholar] [CrossRef] [Green Version]
- Wendler, W.M.; Kremmer, E.; Förster, R.; Winnacker, E.L. Identification of pirin, a novel highly conserved nuclear protein. J. Biol. Chem. 1997, 272, 8482–8489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, H.; Bartlam, M.; Zeng, Q.; Miyatake, H.; Hisano, T.; Miki, K.; Wong, L.L.; Gao, G.F.; Rao, Z. Crystal structure of human pirin: An iron-binding nuclear protein and transcription cofactor. J. Biol. Chem. 2004, 279, 1491–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.; Jia, Z. Structural and biochemical analysis reveal pirins to possess quercetinase activity. J. Biol. Chem. 2005, 280, 28675–28682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurmu, D.; Lu, J.; Johnson, K.A.; Nordlund, P.; Holmgren, A.; Erlandsen, H. The crystal structure of the protein YhaK from Escherichia coli reveals a new subclass of redox sensitive enterobacterial bicupins. Proteins 2009, 74, 18–31. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains and Plasmids | Relevant Genotype/Phenotype | Source |
|---|---|---|
| A. baumannnii | ||
| 98-37-09 | Wild type | [28] |
| 98-37-09 YhaK | 98-37-09 yhaK::EZTn5 | [28] |
| 98-37-09 YhaK pWH1266 | 98-37-09 yhaK::EZTn5; pWH1266 | This study |
| 98-37-09 YhaK pYhaK | 98-37-09 yhaK::EZTn5; pYhaK | This study |
| AB5075-UW | Wild type | [30] |
| AB5075 YhaK | ABUW_0207-119::T26 | [30] |
| AB5075 YhaK pWH1266 | ABUW_0207-119::T26; pWH1266 | This study |
| AB5075 YhaK pYhaK | ABUW_0207-119::T26; pYhaK | This study |
| E. coli | ||
| OneShot® TOP10 | F-mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(araleu)7697 galU galK rpsL (StrR) endA1 nupG | Thermo Fisher |
| Plasmids | ||
| pWH1266 | pBR322 derivative; tetR and ampR | [29] |
| pYhaK | pWH1266 containing 98-37-09 YhaK | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, M.; Chojnacki, M.; Blanchard, C.; Cao, X.; Johnson, W.L.; Flaherty, D.; Dunman, P.M. Genetic Determinants of Acinetobacter baumannii Serum-Associated Adaptive Efflux-Mediated Antibiotic Resistance. Antibiotics 2023, 12, 1173. https://doi.org/10.3390/antibiotics12071173
Young M, Chojnacki M, Blanchard C, Cao X, Johnson WL, Flaherty D, Dunman PM. Genetic Determinants of Acinetobacter baumannii Serum-Associated Adaptive Efflux-Mediated Antibiotic Resistance. Antibiotics. 2023; 12(7):1173. https://doi.org/10.3390/antibiotics12071173
Chicago/Turabian StyleYoung, Mikaeel, Michaelle Chojnacki, Catlyn Blanchard, Xufeng Cao, William L. Johnson, Daniel Flaherty, and Paul M. Dunman. 2023. "Genetic Determinants of Acinetobacter baumannii Serum-Associated Adaptive Efflux-Mediated Antibiotic Resistance" Antibiotics 12, no. 7: 1173. https://doi.org/10.3390/antibiotics12071173