

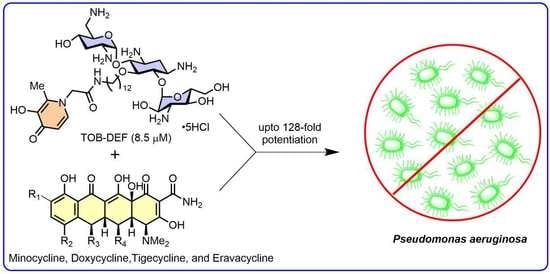
Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa


 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Share and Cite
Gandhi, K.; Dhiman, S.; Arora, R.; Ramirez, D.M.; Ramirez, D.; Arthur, G.; Schweizer, F. Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa. Antibiotics 2023, 12, 1261. https://doi.org/10.3390/antibiotics12081261
Gandhi K, Dhiman S, Arora R, Ramirez DM, Ramirez D, Arthur G, Schweizer F. Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa. Antibiotics. 2023; 12(8):1261. https://doi.org/10.3390/antibiotics12081261
Chicago/Turabian StyleGandhi, Karan, Shiv Dhiman, Rajat Arora, Danzel Marie Ramirez, Danyel Ramirez, Gilbert Arthur, and Frank Schweizer. 2023. "Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa" Antibiotics 12, no. 8: 1261. https://doi.org/10.3390/antibiotics12081261
APA StyleGandhi, K., Dhiman, S., Arora, R., Ramirez, D. M., Ramirez, D., Arthur, G., & Schweizer, F. (2023). Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa. Antibiotics, 12(8), 1261. https://doi.org/10.3390/antibiotics12081261