
Evaluation of the Antibacterial Effect of Aurone-Derived Triazoles on Staphylococcus aureus


Abstract
:1. Introduction
2. Results
2.1. Aurone-Derived Triazole Anti-Staphylococcal Activity and Toxicity for Mammalian Cells
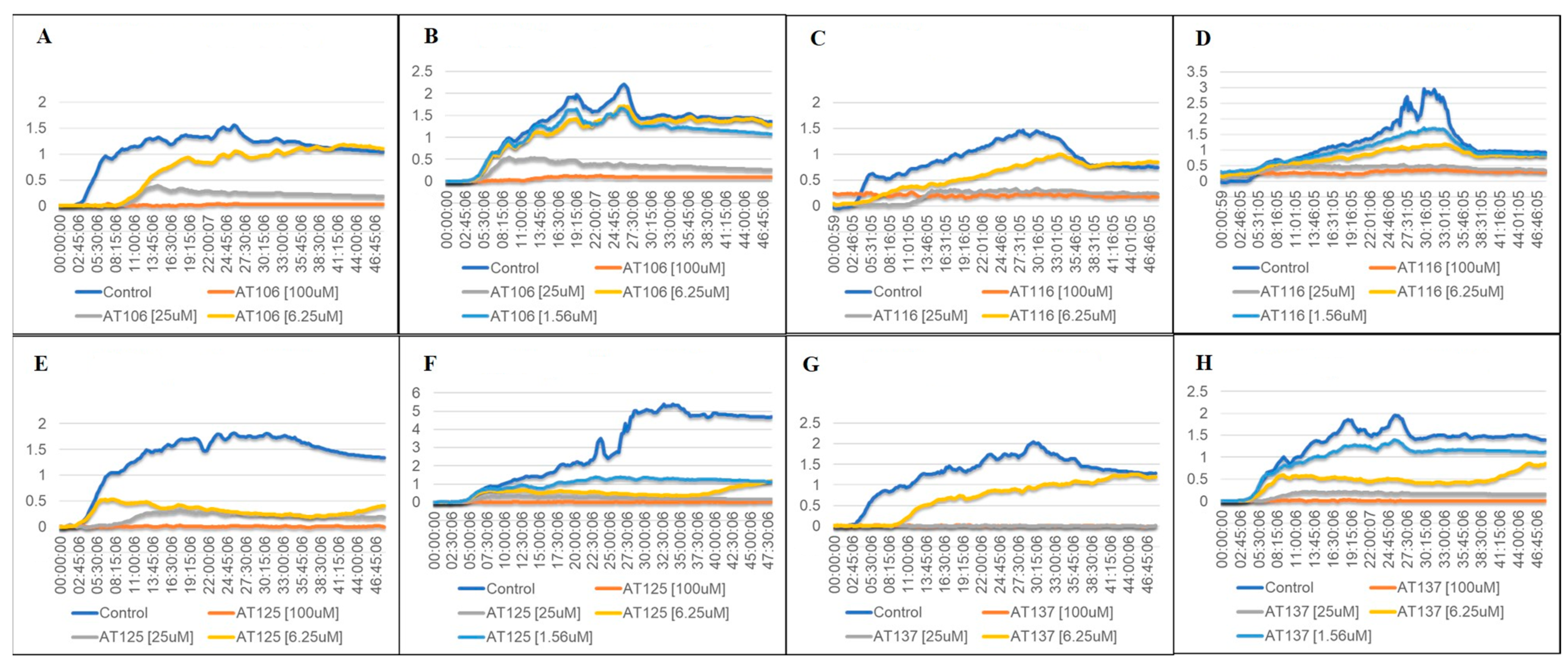
2.2. Bacterial Growth in Aurone-Derived Triazoles
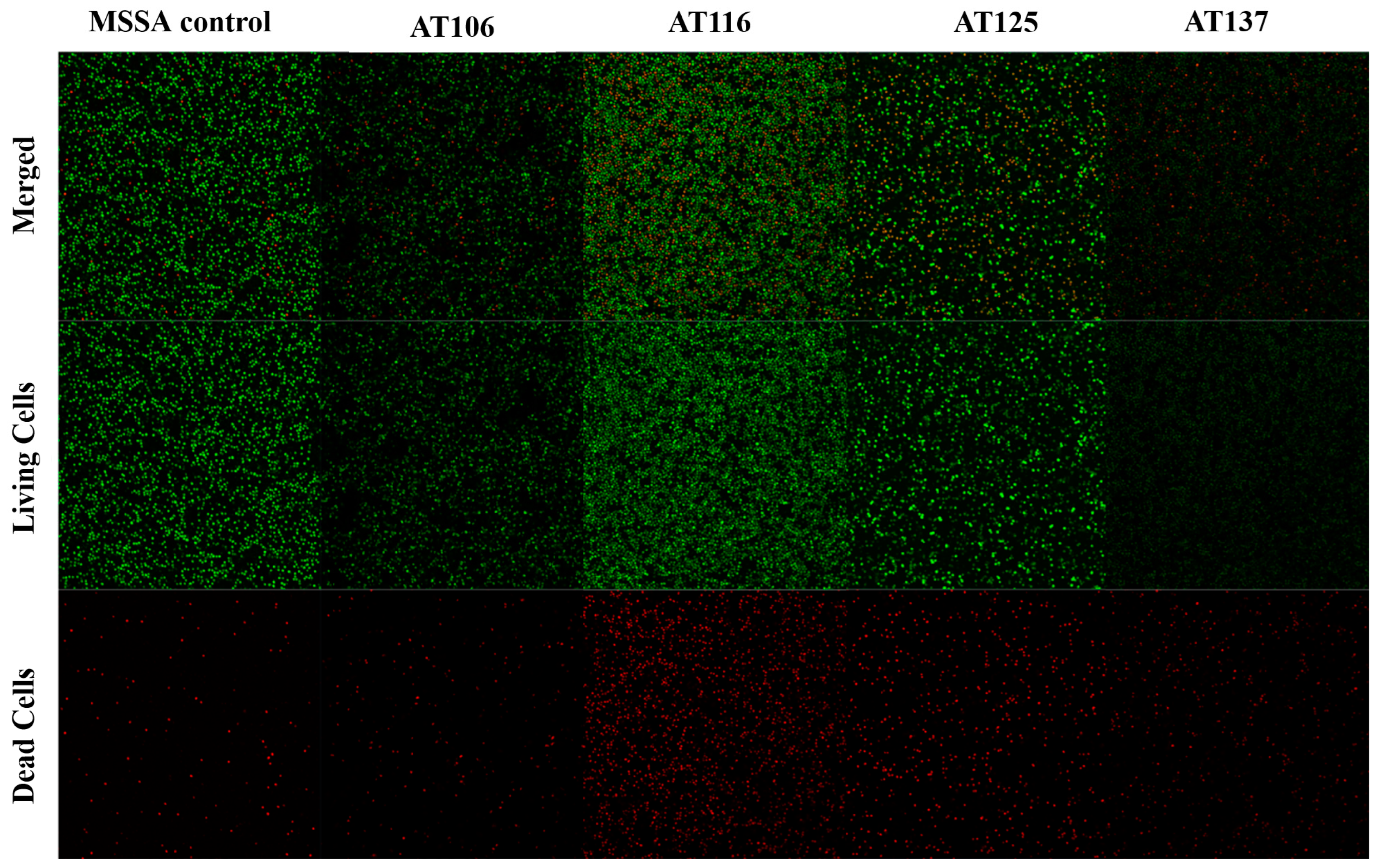
2.3. Biofilm Inhibition Microscopy
2.4. Checkerboard Assay
3. Discussion
4. Materials and Methods
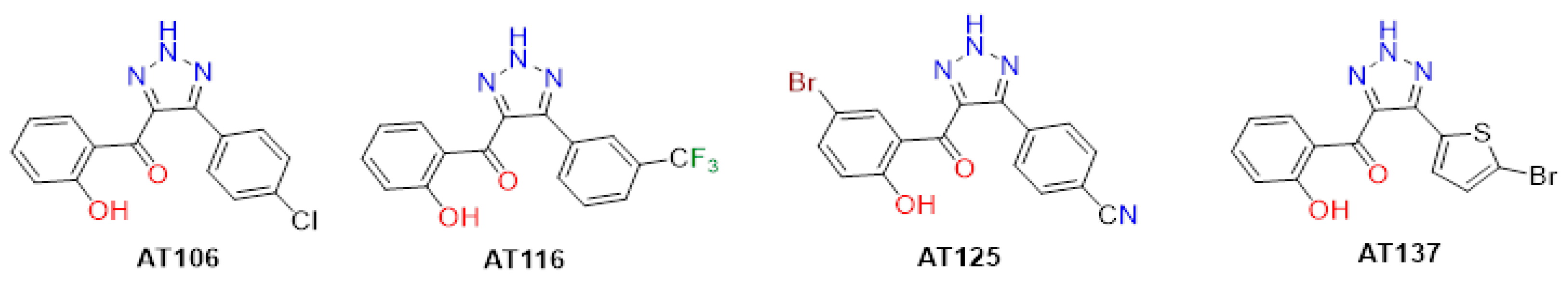
4.1. Aurone-Derived Triazole Compounds
4.2. Bacteria Culture Preparation
4.3. Antibacterial Assay
4.4. Mammalian Cell Lines
4.5. Cytotoxicity Assay
4.6. Hemolysis Assay
4.7. Growth Curve Assay
4.8. Checkerboard Assay
4.9. Biofilm Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Archer, G.L. Staphylococcus aureus: A well-armed pathogen. Clin. Infect. Dis. 1998, 26, 1179–1181. [Google Scholar] [CrossRef]
- Cheung, G.Y.C.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef] [PubMed]
- Kadri, S.S. Key takeaways from the US CDC’s 2019 antibiotic resistance threats report for frontline providers. Crit. Care Med. 2020, 48, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Sharma-Kuinkel, B.T.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Lai, C.F.; Liao, C.H.; Pai, M.F.; Chu, F.Y.; Hsu, S.P.; Chen, H.Y.; Yang, J.Y.; Chiu, Y.L.; Peng, Y.S.; Chang, S.C.; et al. Nasal carriage of methicillin-resistant Staphylococcus aureus is associated with higher all-cause mortality in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 167–174. [Google Scholar] [CrossRef]
- Hanberger, H.; Walther, S.; Leone, M.; Barie, P.S.; Rello, J.; Lipman, J.; Marshall, J.C.; Anzueto, A.; Sakr, Y.; Pickkers, P.; et al. Increased mortality associated with methicillin-resistant Staphylococcus aureus (MRSA) infection in the intensive care unit: Results from the EPIC II study. Int. J. Antimicrob. Agents 2011, 38, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Peacock, S.J.; Paterson, G.K. Mechanisms of methicillin resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef]
- Wulf, M.; Voss, A. MRSA in livestock animals—An epidemic waiting to happen? Clin. Microbiol. Infect. 2008, 14, 519–521. [Google Scholar] [CrossRef]
- Khairullah, A.R.; Kurniawan, S.C.; Effendi, M.H.; Sudjarwo, S.A.; Ramandinianto, S.C.; Widodo, A.; Riwu, K.H.P.; Silaen, O.S.M.; Rehman, S. A review of new emerging livestock-associated methicillin-resistant Staphylococcus aureus from pig farms. Vet. World 2023, 16, 46–58. [Google Scholar] [CrossRef]
- Silva, V.; Monteiro, A.; Pereira, J.E.; Maltez, L.; Igrejas, G.; Poeta, P. MRSA in humans, pets and livestock in Portugal: Where we came from and where we are going. Pathogens 2022, 11, 1110. [Google Scholar] [CrossRef]
- Appelbaum, P.C.; Bozdogan, B. Vancomycin resistance in Staphylococcus aureus. Clin. Lab. Med. 2004, 24, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcal biofilms. Microbiol. Spectr. 2018, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Moormeier, D.E.; Bayles, K.W. Staphylococcus aureus biofilm: A complex developmental organism. Mol. Microbiol. 2017, 104, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Idrees, M.; Sawant, S.; Karodia, N.; Rahman, A. Staphylococcus aureus Biofilm: Morphology, Genetics, Pathogenesis and Treatment Strategies. Int. J. Environ. Res. Public Health 2021, 18, 7602. [Google Scholar] [CrossRef]
- Peng, Q.; Tang, X.; Dong, W.; Sun, N.; Yuan, W. A review of biofilm formation of Staphylococcus aureus and its regulation mechanism. Antibiotics 2023, 12, 12. [Google Scholar] [CrossRef]
- Schilcher, K.; Horswill, A.R. Staphylococcal biofilm development: Structure, regulation, and treatment strategies. Microbiol. Mol. Biol. Rev. 2020, 84, e00026-19. [Google Scholar] [CrossRef]
- Tuon, F.F.; Suss, P.H.; Telles, J.P.; Dantas, L.R.; Borges, N.H.; Ribeiro, V.S.T. Antimicrobial treatment of Staphylococcus aureus biofilms. Antibiotics 2023, 12, 87. [Google Scholar] [CrossRef]
- Livermore, D.M.; British Society for Antimicrobial Chemotherapy Working Party on the Urgent Need: Regenerating Antibacterial Drug Discovery and Development. Discovery research: The scientific challenge of finding new antibiotics. J. Antimicrob. Chemother. 2011, 66, 1941–1944. [Google Scholar] [CrossRef]
- Chahine, E.B.; Dougherty, J.A.; Thornby, K.A.; Guirguis, E.H. Antibiotic approvals in the last decade: Are we keeping up with resistance? Ann. Pharmacother. 2021, 56, 441–462. [Google Scholar] [CrossRef]
- Zimlichman, E.; Henderson, D.; Tamir, O.; Franz, C.; Song, P.; Yamin, C.K.; Keohane, C.; Denham, C.R.; Bates, D.W. Health care–associated infections: A meta-analysis of costs and financial impact on the US health care system. JAMA Intern. Med. 2013, 173, 2039–2046. [Google Scholar] [CrossRef]
- Forrester, J.D.; Maggio, P.M.; Tennakoon, L. Cost of health care–associated infections in the United States. J. Patient Saf. 2022, 18, e477–e479. [Google Scholar] [CrossRef]
- Hübner, C.; Hübner, N.O.; Hopert, K.; Maletzki, S.; Flessa, S. Analysis of MRSA-attributed costs of hospitalized patients in Germany. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1817–1822. [Google Scholar] [CrossRef]
- Macedo-Vinas, M.; De Angelis, G.; Rohner, P.; Safran, E.; Stewardson, A.; Fankhauser, C.; Schrenzel, J.; Pittet, D.; Harbarth, S. Burden of meticillin-resistant Staphylococcus aureus infections at a Swiss university hospital: Excess length of stay and costs. J. Hosp. Infect. 2013, 84, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Gidey, K.; Gidey, M.T.; Hailu, B.Y.; Gebreamlak, Z.B.; Niriayo, Y.L. Clinical and economic burden of healthcare-associated infections: A prospective cohort study. PLoS ONE 2023, 18, e0282141. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Spencer, A.; Long, Y.; Greenhalgh, C.; Steeg, S.; Verma, A. A systematic review and meta-analysis of disease burden of healthcare-associated infections in China: An economic burden perspective from general hospitals. J. Hosp. Infect. 2022, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, I.; Petrarolo, G.; La Motta, C. Aurones: A golden resource for active compounds. Molecules 2021, 27, 2. [Google Scholar] [CrossRef] [PubMed]
- Zwergel, C.; Gaascht, F.; Valente, S.; Diederich, M.; Bagrel, D.; Kirsch, G. Aurones: Interesting natural and synthetic compounds with emerging biological potential. Nat. Prod. Commun. 2012, 7, 389–394. [Google Scholar] [CrossRef]
- Yang, D.; Taylor, Z.E.; Handy, S.; Li, S.; Liu, J.; Stabenow, J.; Zalduondo, L.; Jonsson, C.B.; Altman, E.; Kong, Y. Identification of anti-tuberculosis compounds from aurone analogs. Front. Microbiol. 2020, 11, 1004. [Google Scholar] [CrossRef]
- Olleik, H.; Yahiaoui, S.; Roulier, B.; Courvoisier-Dezord, E.; Perrier, J.; Pérès, B.; Hijazi, A.; Baydoun, E.; Raymond, J.; Boumendjel, A.; et al. Aurone derivatives as promising antibacterial agents against resistant Gram-positive pathogens. Eur. J. Med. Chem. 2019, 165, 133–141. [Google Scholar] [CrossRef]
- Alqahtani, F.M.; Arivett, B.A.; Taylor, Z.E.; Handy, S.T.; Farone, A.L.; Farone, M.B. Chemogenomic profiling to understand the antifungal action of a bioactive aurone compound. PLoS ONE 2019, 11, e0226068. [Google Scholar] [CrossRef]
- Haudecoeur, R.; Ahmed-Belkacem, A.; Yi, W.; Fortuné, A.; Brillet, R.; Belle, C.; Nicolle, E.; Pallier, C.; Pawlotsky, J.M.; Boumendjel, A. Discovery of naturally occurring aurones that are potent allosteric inhibitors of hepatitis C virus RNA-dependent RNA polymerase. J. Med. Chem. 2011, 54, 5395–5402. [Google Scholar] [CrossRef]
- Hawkins, I.; Handy, S.T. Synthesis of aurones under neutral conditions using a deep eutectic solvent. Tetrahedron 2013, 69, 9200–9204. [Google Scholar] [CrossRef]
- Taylor, Z.E.; Handy, S.T. Parallel synthesis of aurones using a homogeneous scavenger. Organics 2023, 4, 51–58. [Google Scholar] [CrossRef]
- Kafle, A.; Bhattarai, S.; Handy, S.T. An unusual triazole synthesis from aurones. Synthesis 2020, 52, 2337–2346. [Google Scholar] [CrossRef]
- Banat, I.M.; De Rienzo, M.A.D.; Quinn, G.A. Microbial biofilms: Biosurfactants as antibiofilm agents. Appl. Microbiol. Biotechnol. 2014, 98, 9915–9929. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Sumran, G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef] [PubMed]
- Matin, M.M.; Matin, P.; Rahman, M.R.; Ben Hadda, T.; Almalki, F.A.; Mahmud, S.; Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and their derivatives: Chemistry, synthesis, and therapeutic applications. Front. Mol. Biosci. 2022, 9, 864286. [Google Scholar] [CrossRef]
- Terreni, M.; Taccani, M.; Pregnolato, M. New antibiotics for multidrug-resistant bacterial strains: Latest research developments and future perspectives. Molecules 2021, 26, 2671. [Google Scholar] [CrossRef]
- Peyton, L.R.; Gallagher, S.; Hashemzadeh, M. Triazole antifungals: A review. Drugs Today 2015, 51, 705–718. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Xiao, J.; Huang, G. Antibacterial activity study of 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2019, 173, 274–281. [Google Scholar] [CrossRef]
- Ge, X.; Xu, Z. 1,2,4-Triazole hybrids with potential antibacterial activity against methicillin-resistant Staphylococcus aureus. Arch. Pharm. 2021, 354, e2000223. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Chen, Z.A.; Shen, Q.K.; Quan, Z.S. Application of triazoles in the structural modification of natural products. J. Enzyme Inhib. Med. Chem. 2021, 36, 1115–1144. [Google Scholar] [CrossRef]
- Gupta, D.; Jain, D.K. Synthesis, antifungal and antibacterial activity of novel 1,2,4-triazole derivatives. J. Adv. Pharm. Technol. Res. 2015, 6, 141–146. [Google Scholar] [CrossRef]
- Paprocka, R.; Wiese-Szadkowska, M.; Kołodziej, P.; Kutkowska, J.; Balcerowska, S.; Bogucka-Kocka, A. Evaluation of biological activity of new 1,2,4-triazole derivatives containing propionic acid moiety. Molecules 2023, 28, 3808. [Google Scholar] [CrossRef] [PubMed]
- Strzelecka, M.; Świątek, P. 1,2,4-Triazoles as important antibacterial agents. Pharmaceuticals 2021, 14, 224. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Deng, C.; Yan, H.; Wang, J.; Liu, K.; Liu, B.S.; Shi, Y.M. 1,2,3-Triazole-containing hybrids with potential antibacterial activity against ESKAPE pathogens. Eur. J. Med. Chem. 2022, 15, 114888. [Google Scholar] [CrossRef]
- El Malah, T.; Nour, H.F.; Satti, A.A.E.; Hemdan, B.A.; El-Sayed, W.A. Design, synthesis, and antimicrobial activities of 1,2,3-triazole glycoside clickamers. Molecules 2020, 25, 790. [Google Scholar] [CrossRef]
- Kant, R.; Kumar, D.; Agarwal, D.; Gupta, R.D.; Tilak, R.; Awasthi, S.K.; Agarwal, A. Synthesis of newer 1,2,3-triazole linked chalcone and flavone hybrid compounds and evaluation of their antimicrobial and cytotoxic activities. Eur. J. Med. Chem. 2016, 113, 34–49. [Google Scholar] [CrossRef]
- Pereira, D.; Pinto, M.; Correia-da-Silva, M.; Cidade, H. Recent advances in bioactive flavonoid hybrids linked by 1,2,3-triazole ring obtained by click chemistry. Molecules 2021, 27, 230. [Google Scholar] [CrossRef]
- Upadhyay, H.C. Coumarin-1,2,3-triazole hybrid molecules: An emerging scaffold for combating drug resistance. Curr. Top. Med. Chem. 2021, 21, 737–752. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 33rd ed.; CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2023. [Google Scholar]
- Diederen, B.M.; van Duijn, I.; Willemse, P.; Kluytmans, J.A. In vitro activity of daptomycin against methicillin-resistant Staphylococcus aureus, including heterogeneously glycopeptide-resistant strains. Antimicrob. Agents Chemother. 2006, 50, 3189–3191. [Google Scholar] [CrossRef]
- Jian, Y.; Lv, H.; Liu, J.; Huang, Q.; Liu, Y.; Liu, Q.; Li, M. Dynamic changes of Staphylococcus aureus susceptibility to vancomycin, teicoplanin, and linezolid in a central teaching hospital in Shanghai, China, 2008–2018. Front. Microbiol. 2020, 11, 908. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, B.; Zhao, H.; Wang, X.; Rao, L.; Ai, W.; Yu, J.; Guo, Y.; Wu, X.; Yu, F.; et al. In vitro activity of vancomycin, teicoplanin, linezolid and daptomycin against methicillin-resistant Staphylococcus aureus isolates collected from Chinese hospitals in 2018–2020. Infect. Drug Resist. 2021, 14, 5449–5456. [Google Scholar] [CrossRef] [PubMed]
- Sederat, Z.; Taylor-Robinson, A.W. Biofilm formation by pathogenic bacteria: Applying a Staphylococcus aureus model to appraise potential targets for therapeutic intervention. Pathogens 2022, 11, 388. [Google Scholar] [CrossRef]
- Sutton, J.A.F.; Carnell, O.T.; Lafage, L.; Gray, J.; Biboy, J.; Gibson, J.F.; Pollitt, E.J.G.; Tazoll, S.C.; Turnbull, W.; Haidamowicz, N.H.; et al. Staphylococcus aureus cell wall structure and dynamics during host-pathogen interaction. PLoS Pathog. 2021, 17, e1009468. [Google Scholar] [CrossRef] [PubMed]
- Frassinetti, S.; Falleni, A.; Del Carratore, R. Effect of itraconazole on Staphylococcus aureus biofilm and extracellular vesicles formation. Microb. Pathog. 2020, 147, 104267. [Google Scholar] [CrossRef] [PubMed]
- Mahey, N.; Tambat, R.; Verma, D.K.; Chandal, N.; Thakur, K.G.; Nandanwar, H. Antifungal azoles as tetracycline resistance modifiers in Staphylococcus aureus. Appl. Environ. Microbiol. 2021, 87, e0015521. [Google Scholar] [CrossRef]
- Kumar, G.; Lathwal, E.; Saroha, B.; Kumar, S.; Kumar, S.; Chauhan, N.S.; Kumar, T. Synthesis and biological evaluation of quinolone-based novel aurones. ChemistrySelect 2020, 5, 3539. [Google Scholar] [CrossRef]
- Sugimoto, S.; Sato, F.; Miyakawa, R.; Chiba, A.; Onodera, S.; Hori, S.; Mizuone, Y. Broad impact of extracellular DNA on biofilm formation by clinically isolated Methicillin-resistant and -sensitive strains of Staphylococcus aureus. Sci. Rep. 2018, 8, 2254. [Google Scholar] [CrossRef]
- Pogliano, J.; Pogliano, N.; Silverman, J.A. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 2012, 194, 4494–4504. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, A.; Almeida, P.F. The antibiotic peptide daptomycin functions by reorganizing the membrane. J. Membr. Biol. 2021, 254, 97–108. [Google Scholar] [CrossRef]
- Cokol, M.; Chua, H.N.; Tasan, M.; Mutlu, B.; Weinstein, Z.B.; Suzuki, Y.; Nergiz, M.E.; Costanzo, M.; Baryshnikova, A.; Giaever, G.; et al. Systematic exploration of synergistic drug pairs. Mol. Syst. Biol. 2011, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Boix-Lemonche, G.; Lekka, M.; Skerlavaj, B. A rapid fluorescence-based microplate assay to investigate the interaction of membrane active antimicrobial peptides with whole gram-positive bacteria. Antibiotics 2020, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; McGoverin, C.; Vanholsbeeck, F.; Swift, S. Optimisation of the protocol for the LIVE/DEAD® BacLightTM Bacterial Viability Kit for rapid determination of bacterial load. Front. Microbiol. 2019, 10, 801. [Google Scholar] [CrossRef] [PubMed]
- Stocks, S.M. Mechanism and use of the commercially available viability stain, BacLight. Cytom. Part A J. Int. Soc. Anal. Cytol. 2004, 61, 189–195. [Google Scholar] [CrossRef]
- Fatsis-Kavalopoulos, N.; Roelofs, L.; Andersson, D.I. Potential risks of treating bacterial infections with a combination of β-lactam and aminoglycoside antibiotics: A systematic quantification of antibiotic interactions in E. coli blood stream infection isolates. EBioMedicine 2022, 78, 103979. [Google Scholar] [CrossRef] [PubMed]
- Lal, J.; Kaul, G.; Akhir, A.; Ansari, S.B.; Chopra, S.; Reddy, D.N. Bio-evaluation of fluoro and trifluoromethyl-substituted salicylanilides against multidrug-resistant S. aureus. Med. Chem. Res. 2021, 30, 2301–2315. [Google Scholar] [CrossRef]
- Gómez, A.C.; Lyons, T.; Mamat, U.; Yero, D.; Bravo, M.; Daura, X.; Elshafee, O.; Brunke, S.; Gahan, C.G.M.; O’Driscoll, M.; et al. Synthesis and evaluation of novel furanones as biofilm inhibitors in opportunistic human pathogens. Eur. J. Med. Chem. 2022, 242, 114678. [Google Scholar] [CrossRef]
- Lonn-Stensrud, J.; Landin, M.A.; Benneche, T.; Petersen, F.C.; Scheie, A.A. Furanones, potential agents for preventing Staphylococcus epidermidis biofilm infections? J. Antimicrob. Chemother. 2009, 63, 309–316. [Google Scholar] [CrossRef]
- Lyons, T.; Gahan, C.G.M.; O’Sullivan, T.P. Structure–activity relationships of furanones, dihydropyrrolones and thiophenones as potential quorum sensing inhibitors. Future Med. Chem. 2020, 12, 1925–1943. [Google Scholar] [CrossRef]
- Steenackers, H.P.; Levin, J.; Janssens, J.C.; De Weerdt, A.; Balzarini, J.; Vanderleyden, J.; De Vos, D.E.; De Keersmaecker, S.C. Structure-activity relationship of brominated 3-alkyl-5-methylene-2(5H)-furanones and alkylmaleic anhydrides as inhibitors of Salmonella biofilm formation and quorum sensing regulated bioluminescence in Vibrio harveyi. Bioorg. Med. Chem. 2010, 18, 5224–5233. [Google Scholar] [CrossRef] [PubMed]
- van Geelen, L.; Kaschani, F.; Sazzadeh, S.S.; Adeniyi, E.T.; Meier, D.; Proksch, P.; Pfeffer, K.; Kaiser, M.; Ioerger, T.R.; Kalscheuer, R. Natural brominated phenoxyphenols kill persistent and biofilm-incorporated cells of MRSA and other pathogenic bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 5985–5998. [Google Scholar] [CrossRef]
- Zang, T.; Lee, B.W.; Cannon, L.M.; Ritter, K.A.; Dai, S.; Ren, D.; Wood, T.K.; Zhou, Z.S. A naturally occurring brominated furanone covalently modifies and inactivates LuxS. Bioorg. Med. Chem. Lett. 2009, 19, 6200–6204. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.L.; Kafle, A.; Handy, S.T.; Farone, A.L.; Miller, J.M. Aurone-derived 1,2,3-triazoles as potential fluorescence molecules in vitro. RSC Adv. 2021, 12, 22639–22649. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Swift, M.L. GraphPad prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. 1997, 37, 411–412. [Google Scholar] [CrossRef]
- Medina, A.; Lambert, R.J.; Magan, N. Rapid throughput analysis of filamentous fungal growth using turbidimetric measurements with the Bioscreen C: A tool for screening antifungal compounds. Fungal Biol. 2012, 116, 161–169. [Google Scholar] [CrossRef]
- Bellio, P.; Fagnani, L.; Nazzicone, L.; Celenza, G. New and simplified method for drug combination studies by checkerboard assay. MethodsX 2021, 8, 101543. [Google Scholar] [CrossRef]
- Rondevaldova, J.; Novy, P.; Urban, J.; Kokoska, L. Determination of anti-staphylococcal activity of thymoquinone in combinations with antibiotics by checkerboard method using EVA capmat™ as a vapor barrier. Arab. J. Chem. 2017, 10, 566–572. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Kapur, J.N.; Sahoo, P.K.; Wong, A.K.C. A new method for gray-level picture thresholding using the entropy of the histogram. Comput. Graph. Image Process. 1985, 29, 273–285. [Google Scholar] [CrossRef]
- Soille, P.; Vincent, L.M. Determining watersheds in digital pictures via flooding simulations. In Proceedings of the Visual Communications and Image Processing ‘90 (Fifth in a Series), Lausanne, Switzerland, 1 September 1990. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| AT | IC50 (µM) | CC50 (µM) | Max SI (CC50/IC50) | ||
|---|---|---|---|---|---|
| MSSA | MRSA | HepG2 | HeLa | ||
| AT106 | 5.439 | 22.380 | 70.660 | 60.330 | 12.99 |
| AT116 | 3.178 | 8.295 | 50.870 | 56.770 | 16.01 |
| AT125 | 4.325 | 5.412 | 50.570 | 51.300 | 11.69 |
| AT137 | 3.092 | 3.870 | 39.810 | 2.838 | 12.88 |
| Concentration (µM) | Hemolysis by AT Compounds (%) | |||
|---|---|---|---|---|
| AT106 | AT116 | AT125 | AT137 | |
| 100 | 1.1 | 3.5 | 5.1 | 1.8 |
| 50 | −0.3 | 2.6 | 4.9 | 1.4 |
| 25 | 1.2 | 2.1 | 3.9 | 2.7 |
| 12.5 | 1.2 | 2.3 | 2.3 | −0.8 |
| 6.25 | −0.1 | −0.7 | 3.6 | −0.5 |
| 3.125 | 0.6 | 0.8 | 0.9 | −2.6 |
| FICi | Vancomycin | Daptomycin | ||
|---|---|---|---|---|
| MSSA | MRSA | MSSA | MRSA | |
| AT106 | 0.57 | 0.52 | 0.25 | 0.05 |
| AT116 | 0.25 | 1.03 | 0.28 | 0.05 |
| AT125 | 0.16 | 1.02 | 1.18 | 0.38 |
| AT137 | 1.5 | 1.13 | 0.38 | 0.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szepe, C.K.; Kafle, A.; Bhattarai, S.; Handy, S.T.; Farone, M.B. Evaluation of the Antibacterial Effect of Aurone-Derived Triazoles on Staphylococcus aureus. Antibiotics 2023, 12, 1370. https://doi.org/10.3390/antibiotics12091370
Szepe CK, Kafle A, Bhattarai S, Handy ST, Farone MB. Evaluation of the Antibacterial Effect of Aurone-Derived Triazoles on Staphylococcus aureus. Antibiotics. 2023; 12(9):1370. https://doi.org/10.3390/antibiotics12091370
Chicago/Turabian StyleSzepe, Csilla Klara, Arjun Kafle, Shrijana Bhattarai, Scott T. Handy, and Mary B. Farone. 2023. "Evaluation of the Antibacterial Effect of Aurone-Derived Triazoles on Staphylococcus aureus" Antibiotics 12, no. 9: 1370. https://doi.org/10.3390/antibiotics12091370