
Data-Driven Approaches in Antimicrobial Resistance: Machine Learning Solutions

 ,
,  , , ,
, , ,  , ,
, , 
Abstract
1. Introduction
- Application of Unsupervised ML Techniques: We leverage K-means clustering and PCA to explore patterns in AMR gene data, offering a novel approach compared to widely used supervised methods.
- Identification of Novel Patterns: Our study uncovers novel patterns in gene length and resistance class that enhance the understanding of the mechanisms underlying AMR.
- Informing Public Health Interventions: We demonstrate the potential of clustering techniques to predict resistance phenotypes, which can inform and guide public health interventions aimed at addressing AMR.
2. Related Work
3. Results
- Pandas: For data manipulation and preprocessing [29]. Pandas provided data structures and functions needed to clean and analyze the dataset efficiently.
- Scikit-learn: For implementing ML algorithms, including K-means clustering and PCA [30]. Scikit-learn is a robust library offering a wide range of ML tools.
3.1. Clustering Outcomes
3.1.1. Cluster Composition
3.1.2. Gene Length Distribution
- Cluster 1: This cluster contained the shortest genes, with a mean length of 493 base pairs (bp) and a standard deviation of 100 bp. The gene lengths ranged from 300 bp to 700 bp, indicating low variability and a tight distribution around the mean.
- Cluster 0: Genes in this cluster were of intermediate length, with a mean of 960 bp and a standard deviation of 141 bp. The lengths ranged from 700 bp to 1200 bp, showing moderate variability.
- Cluster 2: This cluster comprised the longest genes, with a mean length of 1926 bp and a standard deviation of 224 bp. Gene lengths ranged from 1500 bp to 2500 bp, indicating higher variability within the cluster.
3.1.3. Encoded Resistance Class Distribution
- Cluster 1: Predominantly consisted of lower encoded class values, reflecting specific resistance classes associated with simpler mechanisms.
- Cluster 0: Exhibited a moderate range of encoded class values, indicating a diversity of resistance classes.
- Cluster 2: Contained higher encoded class values, corresponding to different resistance classes that may be associated with more complex mechanisms.
3.2. Visualization of Clusters
3.2.1. PCA Scatter Plot
- Cluster 1 (Blue): Positioned towards the lower values of both principal components, reflecting shorter gene lengths and lower encoded class labels.
- Cluster 0 (Red): Occupies an intermediate position between Clusters 1 and 2, indicating moderate gene lengths and a range of encoded class labels.
- Cluster 2 (Green): Located toward higher values of the first principal component, corresponding to longer gene lengths and higher encoded class labels.
3.2.2. Gene Length Distribution by Cluster
- Cluster 1: Displays a narrow distribution with shorter gene lengths, indicating low variability.
- Cluster 0: Shows a moderate distribution of gene lengths, with variability around the median.
- Cluster 2: Exhibits a wider range of longer gene lengths, indicating higher variability within this cluster.
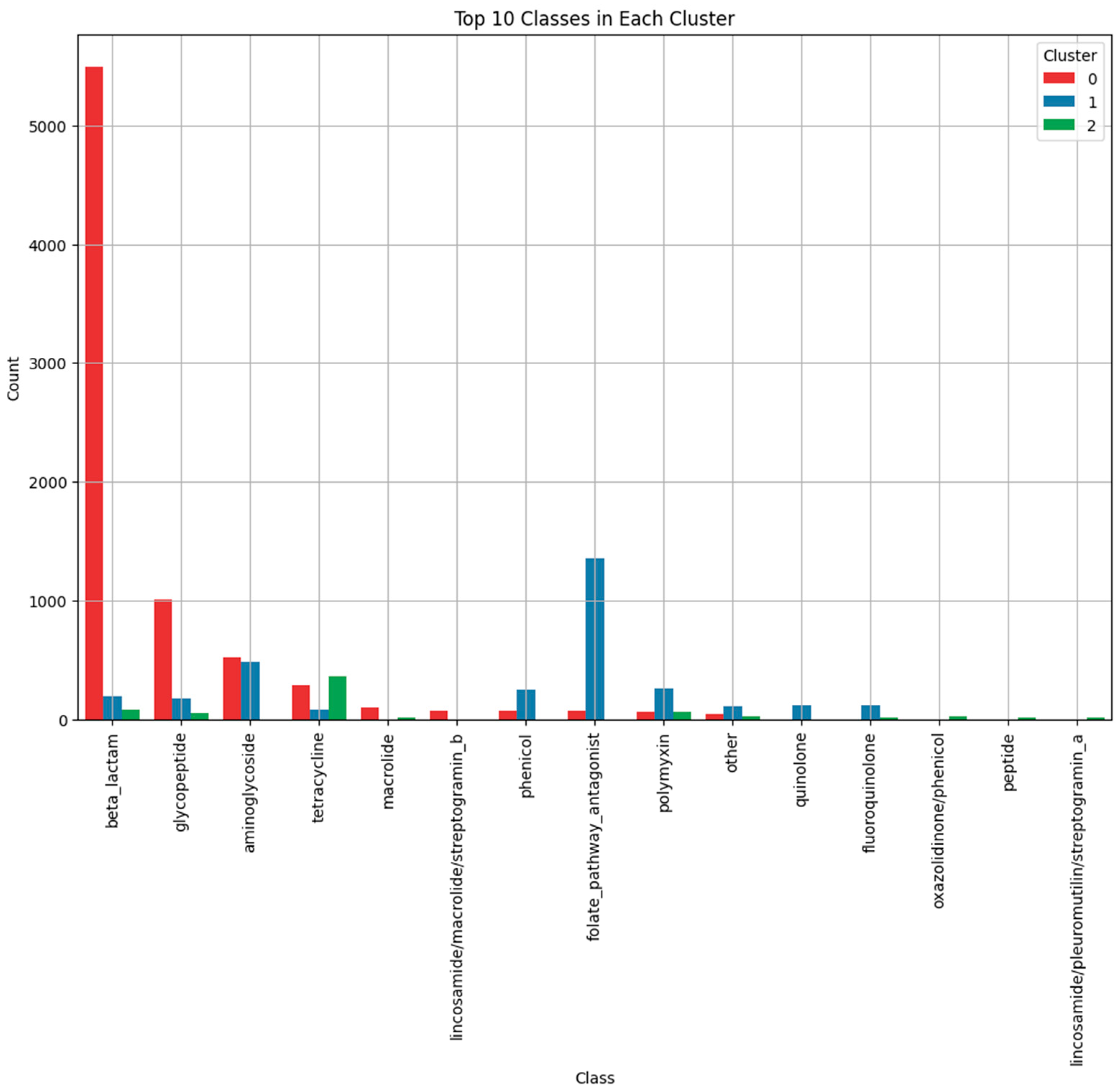
3.2.3. Top Resistance Classes in Each Cluster
- Cluster 0: Dominated by β-lactamase genes and glycopeptide resistance genes. β-lactamases hydrolyze β-lactam antibiotics, rendering them ineffective [36], while glycopeptide resistance involves modification of target sites to prevent antibiotic binding [37]. Genes in this cluster represent a balance between simple and complex resistance mechanisms.
3.3. Statistical Analysis
3.3.1. Analysis of Variance (ANOVA)
- F-statistic: 12,500
- p-value: <0.001
3.3.2. Chi-Square Test for Independence
- Chi-square statistic: 9200
- p-value: <0.001
3.4. Biological Interpretation of Clustering Results
3.4.1. Correlation Between Gene Length and Resistance Mechanisms
- Shorter Genes (Cluster 1): Genes in this cluster are shorter and associated with simpler resistance mechanisms, such as antibiotic inactivation or metabolic pathway bypass [43]. For example, sul1 and sul2 confer resistance to sulfonamides by encoding dihydropteroate synthase variants that are less sensitive to inhibition [34]. Cat genes encode chloramphenicol acetyltransferases that inactivate chloramphenicol [35].
- Intermediate-Length Genes (Cluster 0): These genes are associated with mechanisms like antibiotic degradation and target modification. β-lactamases hydrolyze the β-lactam ring of antibiotics, neutralizing their antibacterial activity [36]. Glycopeptide resistance involves the alteration of cell wall precursors, preventing the binding of antibiotics like vancomycin [37].
- Longer Genes (Cluster 2): Genes in this cluster are longer and linked to more complex resistance mechanisms requiring larger protein structures. Aminoglycoside-modifying enzymes (e.g., aac(6′)-Ib) modify the antibiotic, reducing its affinity for the target [38]. Tetracycline resistance genes (e.g., tet(M)) encode ribosomal protection proteins that prevent tetracycline from binding to the ribosome [39]. Efflux pumps actively transport antibiotics out of the cell, a mechanism that often involves large transmembrane proteins [44].
3.4.2. Implications for Horizontal Gene Transfer
- Longer genes (Cluster 2) may be less frequently transferred via HGT due to size and energy constraints but can still spread via integrative conjugative elements and bacteriophages [45].
- Understanding the link between gene length and mobility can help in controlling AMR gene dissemination in clinical and environmental settings.
3.4.3. Potential Identification of Novel Resistance Mechanisms
4. Discussion
4.1. Interpretation of Findings
4.2. Clinical and Public Health Implications
4.3. Contributions to AMR Research
4.4. Limitations of the Study
4.5. Future Directions
4.6. Ethical and Societal Considerations
5. Methods
5.1. Dataset Overview
5.2. Data Cleaning and Preprocessing
5.2.1. Filtering for Relevant Resistance Types
5.2.2. Removal of Irrelevant Columns
5.2.3. Handling Missing Values
5.2.4. Encoding Categorical Variables
5.2.5. Calculation of Gene Lengths
5.2.6. Data Normalization
5.2.7. Dimensionality Reduction Preparation
5.2.8. Exploratory Data Analysis (EDA)
5.2.9. Validation of Data Integrity
5.2.10. Final Dataset Composition
5.3. Feature Selection for Clustering
5.4. K-Means Clustering
5.4.1. Rationale for Algorithm Selection
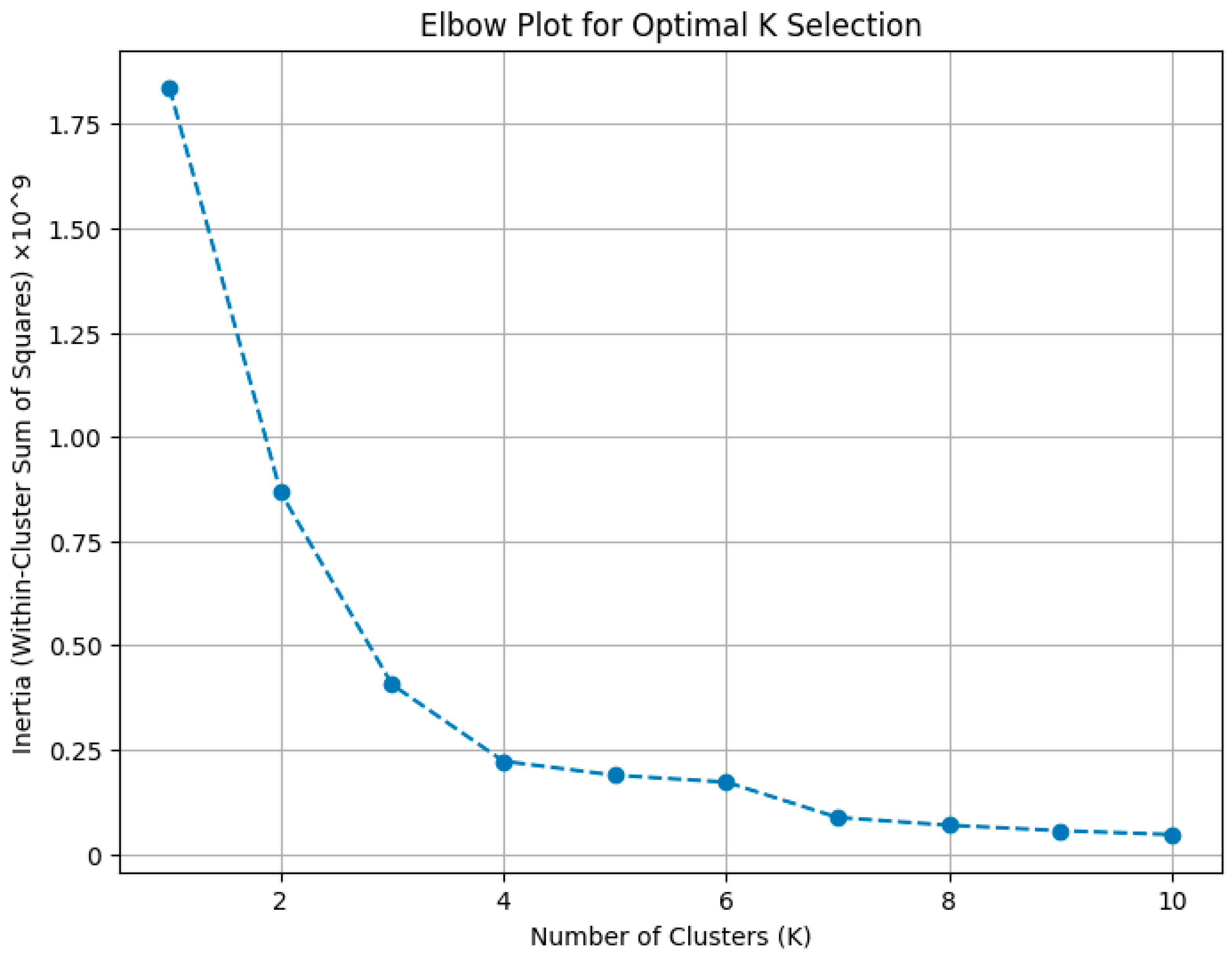
5.4.2. Determining the Optimal Number of Clusters
5.4.3. Clustering Procedure
- Initialization: Centroids were initialized randomly, and a fixed random state was set to ensure reproducibility of the results.
- Iteration: The algorithm iteratively assigned each data point to the nearest centroid based on Euclidean distance and then recalculated the centroids as the mean position of all points assigned to each cluster.
- Convergence: The process continued until the centroids no longer shifted significantly between iterations, indicating that the clusters had stabilized.
- Cluster Assignment: Each gene was assigned a cluster label (0, 1, or 2), corresponding to one of the three clusters identified.
5.5. Principal Component Analysis
5.6. Statistical Analysis and Visualization
5.6.1. Descriptive Statistics
- Cluster Counts: The number of genes in each cluster was calculated to assess the distribution of data points among clusters.
- Gene Length Statistics: For each cluster, the mean, median, variance, standard deviation, minimum, and maximum gene lengths were calculated. These statistics provided insights into the central tendencies and variability of gene lengths within clusters.
- Resistance Class Statistics: The mean and median of the encoded resistance classes were computed to understand the distribution of resistance types within each cluster.
5.6.2. Data Visualization
- PCA Scatter Plot: A scatter plot of the first two principal components was created to visualize the clustering of data points in two dimensions. Data points were colored according to their assigned clusters, allowing for visual assessment of cluster separation.
- Boxplots of Gene Lengths: Boxplots were generated to display the distribution of gene lengths within each cluster. This visualization highlighted differences in gene length distributions among clusters.
- Bar Plots of Top Resistance Classes: Bar plots were created to showcase the top 10 most frequent resistance classes within each cluster. This helped identify dominant resistance mechanisms associated with each cluster.
- Pie Charts of Class Distribution: Pie charts illustrate the proportion of different resistance classes within each cluster, providing a visual representation of class diversity.
5.6.3. Statistical Analysis Methods
- Analysis of Variance (ANOVA): ANOVA was used to determine if there were statistically significant differences in gene lengths among the clusters [40]. A significant F-test would indicate that at least one cluster's mean gene length is different from the others.
- Chi-Square Test for Independence: A chi-square test was performed to assess the association between clusters and resistance classes [42]. A significant result would suggest that the distribution of resistance classes is not independent of cluster assignment.
5.7. Reproducibility and Code Availability
5.8. Ethical Considerations
5.9. Justification of Methodological Choices
- Unsupervised Learning: Given the exploratory nature of the study and the lack of predefined labels for grouping, unsupervised learning methods like K-means clustering were appropriate.
- Dimensionality Reduction: PCA was necessary to visualize the data effectively and to identify underlying patterns that are not apparent in higher dimensions.
- Statistical Analysis: Employing statistical tests ensured that the observed patterns and differences were not due to random chance, adding trust to the findings.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Antimicrobial Resistance. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 17 August 2024).
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance. 2016. Available online: https://wellcomecollection.org/works/thvwsuba (accessed on 17 August 2024).
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar] [CrossRef] [PubMed]
- von Wintersdorff, C.J.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.; Wolffs, P.F. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Wright, G.D. The antibiotic resistance “mobilome”: Searching for the link between environment and clinic. Front. Microbiol. 2013, 4, 138. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Sifakis, F.; Harbarth, S.; Schrijver, R.; van Mourik, M.; Voss, A.; Sharland, M.; Rajendran, N.B.; Rodríguez-Baño, J.; EPI-Net COMBACTE-MAGNET Group. Surveillance for control of antimicrobial resistance. Lancet Infect. Dis. 2018, 18, e99–e106. [Google Scholar] [CrossRef] [PubMed]
- van Belkum, A.; Bachmann, T.T.; Lüdke, G.; Lisby, J.G.; Kahlmeter, G.; Mohess, A.; Becker, K.; Hays, J.P.; Woodford, N.; Mitsakakis, K.; et al. Developmental roadmap for antimicrobial susceptibility testing systems. Nat. Rev. Microbiol. 2019, 17, 51–62. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 4694, 967–977. [Google Scholar] [CrossRef]
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef]
- Jordan, M.I.; Mitchell, T.M. Machine learning: Trends, perspectives, and prospects. Science 2015, 349, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Anahtar, M.N.; Yang, J.H.; Kanjilal, S. Applications of Machine Learning to the Problem of Antimicrobial Resistance: An Emerging Model for Translational Research. J. Clin. Microbiol. 2021, 59, e0126020. [Google Scholar] [CrossRef] [PubMed]
- Feretzakis, G.; Loupelis, E.; Sakagianni, A.; Kalles, D.; Lada, M.; Christopoulos, C.; Dimitrellos, E.; Martsoukou, M.; Skarmoutsou, N.; Petropoulou, S.; et al. Using machine learning algorithms to predict antimicrobial resistance and assist empirical treatment. Stud. Health Technol. Inform. 2020, 272, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Sakagianni, A.; Feretzakis, G.; Kalles, D.; Loupelis, E.; Rakopoulou, Z.; Dalainas, I.; Fildisis, G. Discovering Association Rules in Antimicrobial Resistance in Intensive Care Unit. Stud. Health Technol. Inform. 2022, 295, 430–433. [Google Scholar] [CrossRef]
- Mahé, P.; Tournoud, M. Predicting bacterial resistance from whole-genome sequences using k-mers and stability selection. BMC Bioinform. 2018, 19, 383. [Google Scholar] [CrossRef]
- Nguyen, M.; Long, S.W.; McDermott, P.F.; Olsen, R.J.; Olson, R.; Stevens, R.L.; Tyson, G.H.; Zhao, S.; Davis, J.J. Using Machine Learning To Predict Antimicrobial MICs and Associated Genomic Features for Nontyphoidal Salmonella. J. Clin. Microbiol. 2019, 57, e01260-18. [Google Scholar] [CrossRef]
- Kotwal, S.; Rani, P.; Arif, T.; Manhas, J.; Sharma, S. Automated Bacterial Classifications Using Machine Learning Based Computational Techniques: Architectures, Challenges and Open Research Issues. Arch. Comput. Methods Eng. State Art Rev. 2022, 29, 2469–2490. [Google Scholar] [CrossRef]
- Branda, F.; Scarpa, F. Implications of Artificial Intelligence in Addressing Antimicrobial Resistance: Innovations, Global Challenges, and Healthcare’s Future. Antibiotics 2024, 13, 502. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Martiny, H.-M.; Pyrounakis, N.; Lukjančenko, O.; Petersen, T.N.; Aarestrup, F.M.; Clausen, P.T.L.C.; Munk, P. PanRes—Collection of Antimicrobial Resistance Genes (1.0.0) [Data Set]. Zenodo. 2023. Available online: https://zenodo.org/records/8055116 (accessed on 24 August 2024).
- Boolchandani, M.; D’Souza, A.W.; Dantas, G. Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 2019, 20, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Vellido, A. The importance of interpretability and visualization in machine learning for applications in medicine and health care. Neural Comput. Appl. 2019, 32, 18069–18083. [Google Scholar] [CrossRef]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Char, D.S.; Shah, N.H.; Magnus, D. Implementing Machine Learning in Health Care—Addressing Ethical Challenges. N. Engl. J. Med. 2018, 378, 981–983. [Google Scholar] [CrossRef]
- Yang, Y.; Niehaus, K.E.; Walker, T.M.; Iqbal, Z.; Walker, A.S.; Wilson, D.J.; Peto, T.E.A.; Crook, D.W.; Smith, E.G.; Zhu, T.; et al. Machine learning for classifying tuberculosis drug-resistance from DNA sequencing data. Bioinformatics 2018, 34, 1666–1671. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 56–61. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.; Botvinnik, O.; O’Kane, D.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Cole, J.B.; Warmenhoven, J.; et al. mwaskom/seaborn: v0.8.1 (September 2017). Zenodo. 2017. Available online: https://zenodo.org/records/883859 (accessed on 24 August 2024).
- Shutaywi, M.; Kachouie, N.N. Silhouette analysis for performance evaluation in machine learning with applications to clustering. Entropy 2021, 23, 759. [Google Scholar] [CrossRef]
- Sköld, O. Sulfonamide resistance: Mechanisms and trends. Drug Resist. Updates 2000, 3, 155–160. [Google Scholar] [CrossRef]
- Schwarz, S.; Kehrenberg, C.; Doublet, B.; Cloeckaert, A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 2004, 28, 519–542. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Arthur, M.; Courvalin, P. Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob. Agents Chemother. 1993, 37, 1563–1571. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updates 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Connell, S.R.; Tracz, D.M.; Nierhaus, K.H.; Taylor, D.E. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob. Agents Chemother. 2003, 47, 3675–3681. [Google Scholar] [CrossRef]
- Montgomery, D.C. Design and Analysis of Experiments; John Wiley & Sons: Hoboken, NJ, USA, 2017; Available online: https://books.google.gr/books?id=Py7bDgAAQBAJ (accessed on 24 August 2024).
- Abdi, H.; Williams, L.J. Tukey’s honestly significant difference (HSD) test. In Encyclopedia of Research Design; SAGE Publications: New York, NJ, USA, 2010; pp. 1–5. [Google Scholar]
- Agresti, A. An Introduction to Categorical Data Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Poole, K. Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 2007, 39, 162–176. [Google Scholar] [CrossRef]
- Wozniak, R.A.; Waldor, M.K. Integrative and conjugative elements: Mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010, 8, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2020, 48, D561–D569. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Nikaido, H.; Pagès, J.M. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 2012, 36, 340–363. [Google Scholar] [CrossRef]
- San Millan, A. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol. 2018, 26, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.A. Extended-spectrum beta-lactamases in the 21st century: Characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 2001, 14, 933–951. [Google Scholar] [CrossRef] [PubMed]
- Bahl, M.I.; Hansen, L.H.; Sørensen, S.J. Impact of conjugal transfer on the stability of IncP-1 plasmid pKJK5 in bacterial populations. FEMS Microbiol. Lett. 2007, 266, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.P.; Mullany, P. Tn916-like genetic elements: A diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 856–871. [Google Scholar] [CrossRef]
- Martínez, J.L.; Baquero, F. Interactions among strategies associated with bacterial infection: Pathogenicity, epidemicity, and antibiotic resistance. Clin. Microbiol. Rev. 2002, 15, 647–679. [Google Scholar] [CrossRef]
- Livermore, D.M. beta-Lactamases in laboratory and clinical resistance. Clin. Microbiol. Rev. 1995, 8, 557–584. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef]
- Roberts, M.C. Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 2005, 245, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Su, M.; Satola, S.W.; Read, T.D. Genome-Based Prediction of Bacterial Antibiotic Resistance. J. Clin. Microbiol. 2019, 57, e01405-18. [Google Scholar] [CrossRef]
- Dyar, O.J.; Huttner, B.; Schouten, J.; Pulcini, C.; ESGAP (ESCMID Study Group for Antimicrobial stewardshiP). What is antimicrobial stewardship? Clin. Microbiol. Infect. 2017, 23, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.H.; Moore, L.S.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.N.; et al. Tackling antibiotic resistance: The environmental framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Naccache, S.N. Metagenomics to Assist in the Diagnosis of Bloodstream Infection. J. Appl. Lab. Med. 2019, 3, 643–653. [Google Scholar] [CrossRef]
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Sommer, M.O.; Dantas, G. The shared antibiotic resistome of soil bacteria and human pathogens. Science 2012, 337, 1107–1111. [Google Scholar] [CrossRef]
- Mahfouz, N.; Ferreira, I.; Beisken, S.; von Haeseler, A.; Posch, A.E. Large-scale assessment of antimicrobial resistance marker databases for genetic phenotype prediction: A systematic review. J. Antimicrob. Chemother. 2020, 75, 3099–3108. [Google Scholar] [CrossRef]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, X.; Li, J.; Lv, N.; Liu, F.; Wu, J.; Lin, I.Y.; Wu, N.; Weimer, B.C.; Gao, G.F.; et al. The Bacterial Mobile Resistome Transfer Network Connecting the Animal and Human Microbiomes. Appl. Environ. Microbiol. 2016, 82, 6672–6681. [Google Scholar] [CrossRef]
- Steinley, D. K-means clustering: A half-century synthesis. Br. J. Math. Stat. Psychol. 2006, 59, 1–34. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Antibiotic resistance and its cost: Is it possible to reverse resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, Portland, ON, USA, 2–4 August 1996; pp. 226–231. [Google Scholar]
- Fraley, C.; Raftery, A.E. Model-based clustering, discriminant analysis, and density estimation. J. Am. Stat. Assoc. 2002, 97, 611–631. [Google Scholar] [CrossRef]
- Hennig, C. Cluster-wise assessment of cluster stability. Comput. Stat. Data Anal. 2007, 52, 258–271. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Huang, S.; Chaudhary, K.; Garmire, L.X. More is better: Recent progress in multi-omics data integration methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef]
- Recker, M.; Laabei, M.; Toleman, M.S.; Reuter, S.; Saunderson, R.B.; Blane, B.; Torok, M.E.; Ouadi, K.; Stevens, E.; Yokoyama, M.; et al. Clonal differences in Staphylococcus aureus bacteraemia-associated mortality. Nat. Microbiol. 2017, 2, 1381–1388. [Google Scholar] [CrossRef]
- Feretzakis, G.; Sakagianni, A.; Loupelis, E.; Kalles, D.; Skarmoutsou, N.; Martsoukou, M.; Christopoulos, C.; Lada, M.; Petropoulou, S.; Velentza, A.; et al. Machine Learning for Antibiotic Resistance Prediction: A Prototype Using Off-the-Shelf Techniques and Entry-Level Data to Guide Empiric Antimicrobial Therapy. Healthc. Inform. Res. 2021, 27, 214–221. [Google Scholar] [CrossRef]
- Sakagianni, A.; Koufopoulou, C.; Feretzakis, G.; Kalles, D.; Verykios, V.S.; Myrianthefs, P.; Fildisis, G. Using Machine Learning to Predict Antimicrobial Resistance—A Literature Review. Antibiotics 2023, 12, 452. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Hazel, J.W.; Clayton, E.W.; Vorobeychik, Y.; Kantarcioglu, M.; Malin, B.A. Sociotechnical safeguards for genomic data privacy. Nat. Rev. Genet. 2022, 23, 429–445. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Action Plan: On Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2015; pp. I–IV. Available online: http://www.jstor.org/stable/resrep47928.1 (accessed on 2 September 2024).
- Robinson, T.P.; Bu, D.P.; Carrique-Mas, J.; Fèvre, E.M.; Gilbert, M.; Grace, D.; Hay, S.I.; Jiwakanon, J.; Kakkar, M.; Kariuki, S.; et al. Antibiotic resistance is the quintessential One Health issue. Trans. R. Soc. Trop. Med. Hyg. 2016, 110, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Kotsiantis, S.; Kanellopoulos, D.; Pintelas, P. Data preprocessing for supervised learning. Int. J. Comput. Sci. 2006, 1, 111–117. [Google Scholar]
- Martiny, H.-M.; Pyrounakis, N.; Petersen, T.N.; Lukjančenko, O.; Aarestrup, F.M.; Clausen, P.T.L.C.; Munk, P. ARGprofiler—A pipeline for large-scale analysis of antimicrobial resistance genes and their flanking regions in metagenomic datasets. Bioinformatics 2024, 40, btae086. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genom. 2015, 16, 964. [Google Scholar] [CrossRef]
- Batista, G.E.; Monard, M.C. An analysis of four missing data treatment methods for supervised learning. Appl. Artif. Intell. 2003, 17, 519–533. [Google Scholar] [CrossRef]
- Zhang, Z. Missing data imputation: Focusing on single imputation. Ann. Transl. Med. 2016, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K. Data clustering: 50 years beyond K-means. Pattern Recognit. Lett. 2010, 31, 651–666. [Google Scholar] [CrossRef]
- Lopes, I.; Altab, G.; Raina, P.; de Magalhães, J.P. Gene Size Matters: An Analysis of Gene Length in the Human Genome. Front. Genet. 2021, 12, 559998. [Google Scholar] [CrossRef]
- Han, J.; Kamber, M.; Pei, J. Mining frequent patterns, associations, and correlations: Basic concepts and methods. In Data Mining, 3rd ed.; Han, J., Kamber, M., Pei, J., Eds.; Morgan Kaufmann: Burlington, MA, USA, 2012; pp. 243–278. [Google Scholar] [CrossRef]
- Patro, S.G.K.; Sahu, K.K. Normalization: A preprocessing stage. arXiv 2015, arXiv:1503.06462. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Tukey, J.W. Exploratory Data Analysis; Addison-Wesley: Boston, MA, USA, 1977. [Google Scholar]
- Li, H.; Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinform. 2010, 11, 473–483. [Google Scholar] [CrossRef]
- Ikotun, A.M.; Ezugwu, A.E.; Abualigah, L.; Abuhaija, B.; Heming, J. K-means clustering algorithms: A comprehensive review, variants analysis, and advances in the era of big data. Inf. Sci. 2023, 622, 178–210. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Gene_Length | Resistance_Type |
|---|---|---|
| tetracycline | 1233 | antimicrobial |
| glycopeptide | 981 | antimicrobial |
| folate_pathway_antagonist | 561 | antimicrobial |
| glycopeptide | 1056 | antimicrobial |
| folate_pathway_antagonist | 528 | antimicrobial |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakagianni, A.; Koufopoulou, C.; Koufopoulos, P.; Kalantzi, S.; Theodorakis, N.; Nikolaou, M.; Paxinou, E.; Kalles, D.; Verykios, V.S.; Myrianthefs, P.; et al. Data-Driven Approaches in Antimicrobial Resistance: Machine Learning Solutions. Antibiotics 2024, 13, 1052. https://doi.org/10.3390/antibiotics13111052
Sakagianni A, Koufopoulou C, Koufopoulos P, Kalantzi S, Theodorakis N, Nikolaou M, Paxinou E, Kalles D, Verykios VS, Myrianthefs P, et al. Data-Driven Approaches in Antimicrobial Resistance: Machine Learning Solutions. Antibiotics. 2024; 13(11):1052. https://doi.org/10.3390/antibiotics13111052
Chicago/Turabian StyleSakagianni, Aikaterini, Christina Koufopoulou, Petros Koufopoulos, Sofia Kalantzi, Nikolaos Theodorakis, Maria Nikolaou, Evgenia Paxinou, Dimitris Kalles, Vassilios S. Verykios, Pavlos Myrianthefs, and et al. 2024. "Data-Driven Approaches in Antimicrobial Resistance: Machine Learning Solutions" Antibiotics 13, no. 11: 1052. https://doi.org/10.3390/antibiotics13111052
APA StyleSakagianni, A., Koufopoulou, C., Koufopoulos, P., Kalantzi, S., Theodorakis, N., Nikolaou, M., Paxinou, E., Kalles, D., Verykios, V. S., Myrianthefs, P., & Feretzakis, G. (2024). Data-Driven Approaches in Antimicrobial Resistance: Machine Learning Solutions. Antibiotics, 13(11), 1052. https://doi.org/10.3390/antibiotics13111052