
Origami of KR-12 Designed Antimicrobial Peptides and Their Potential Applications

 ,
, 

Abstract
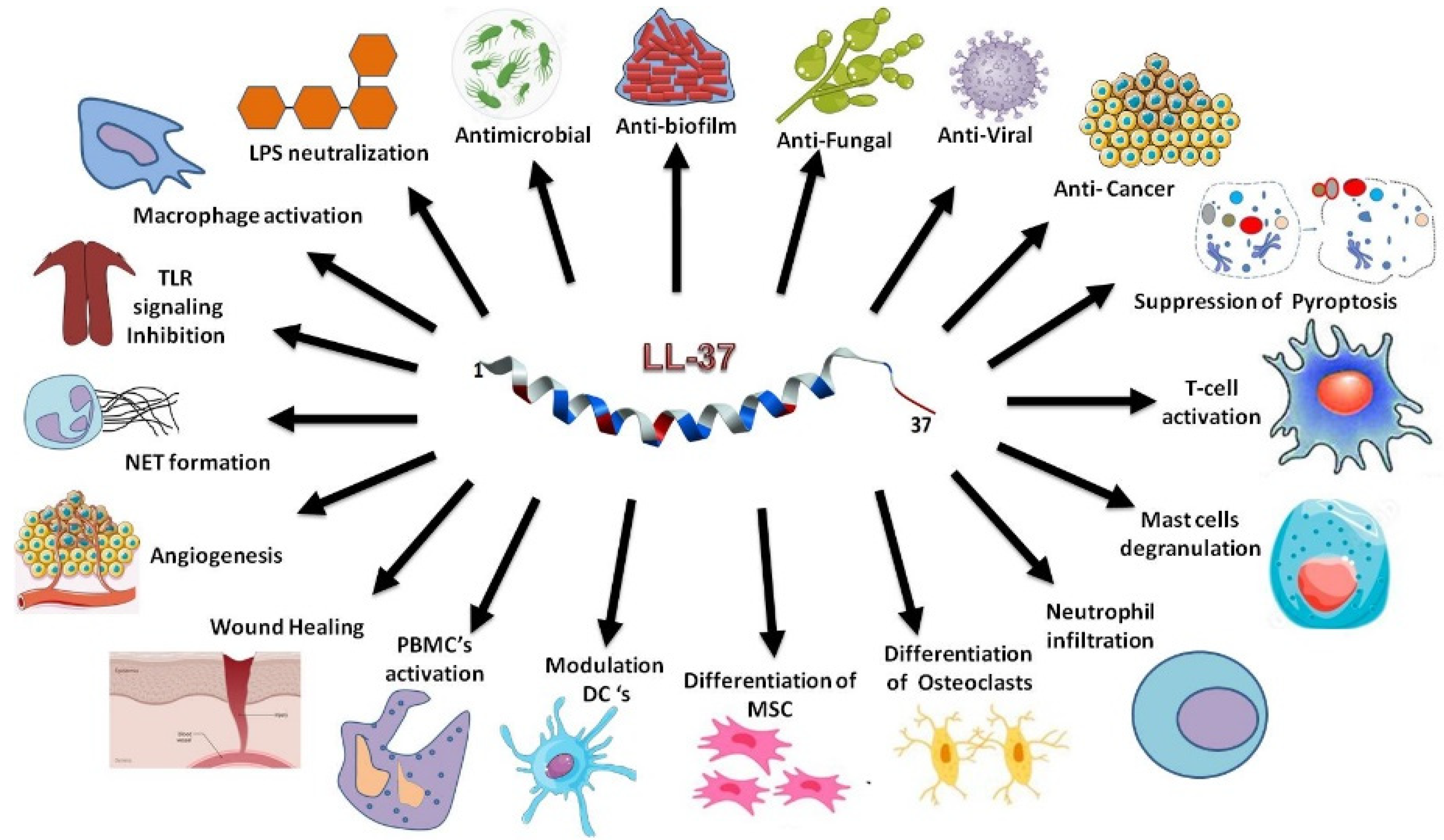
:1. Introduction
2. Discovery, NMR Structure, and Mechanism of Action of KR-12 Derived from the Antimicrobial Core of Human LL-37
2.1. The Discovery of KR-12
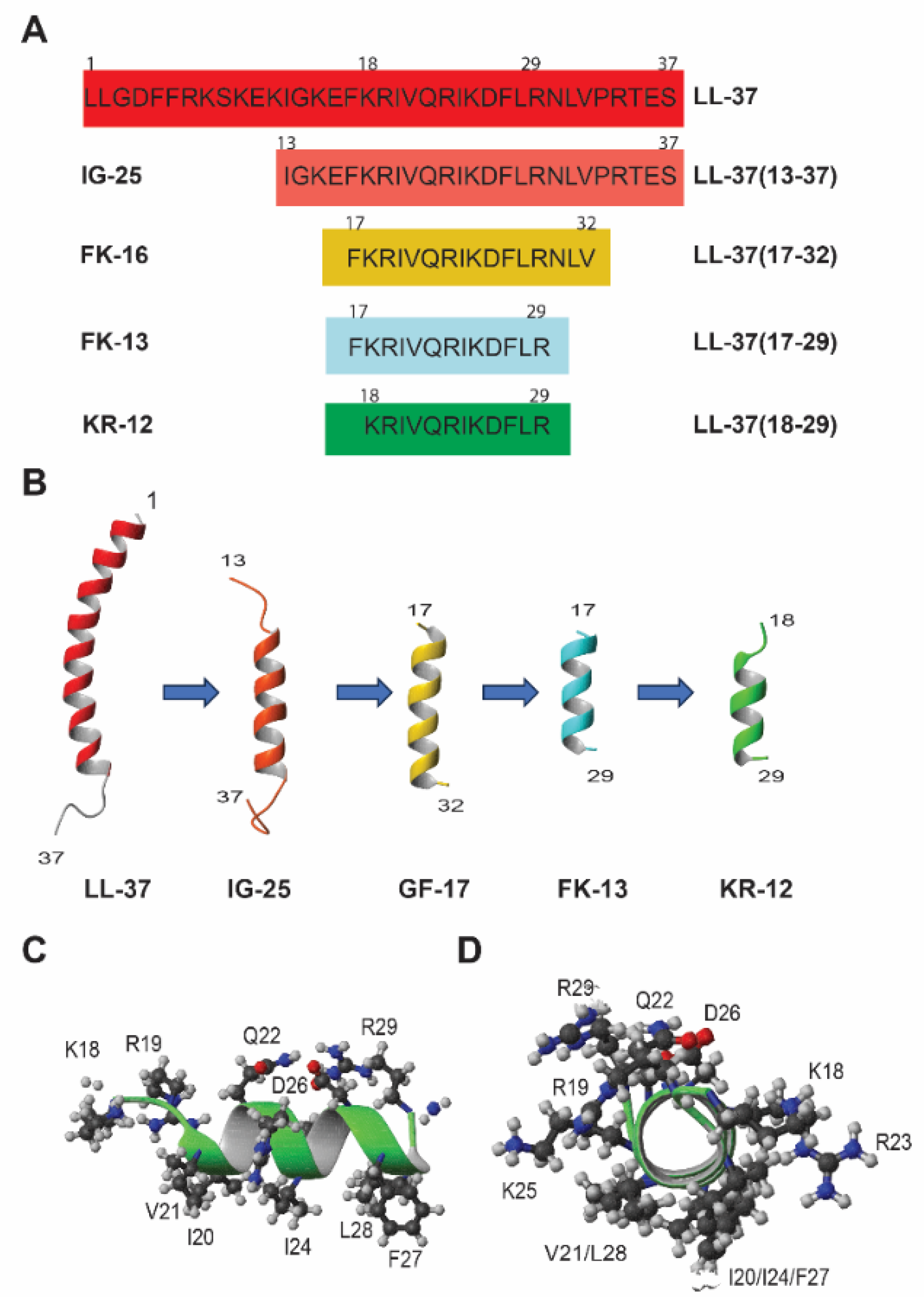
2.2. Structural Determination and Mechanistic Studies of KR-12
3. Structure–Activity Relationship (SAR) of KR-12
3.1. Molecular Forms and Media Conditions Influence Peptide Activity
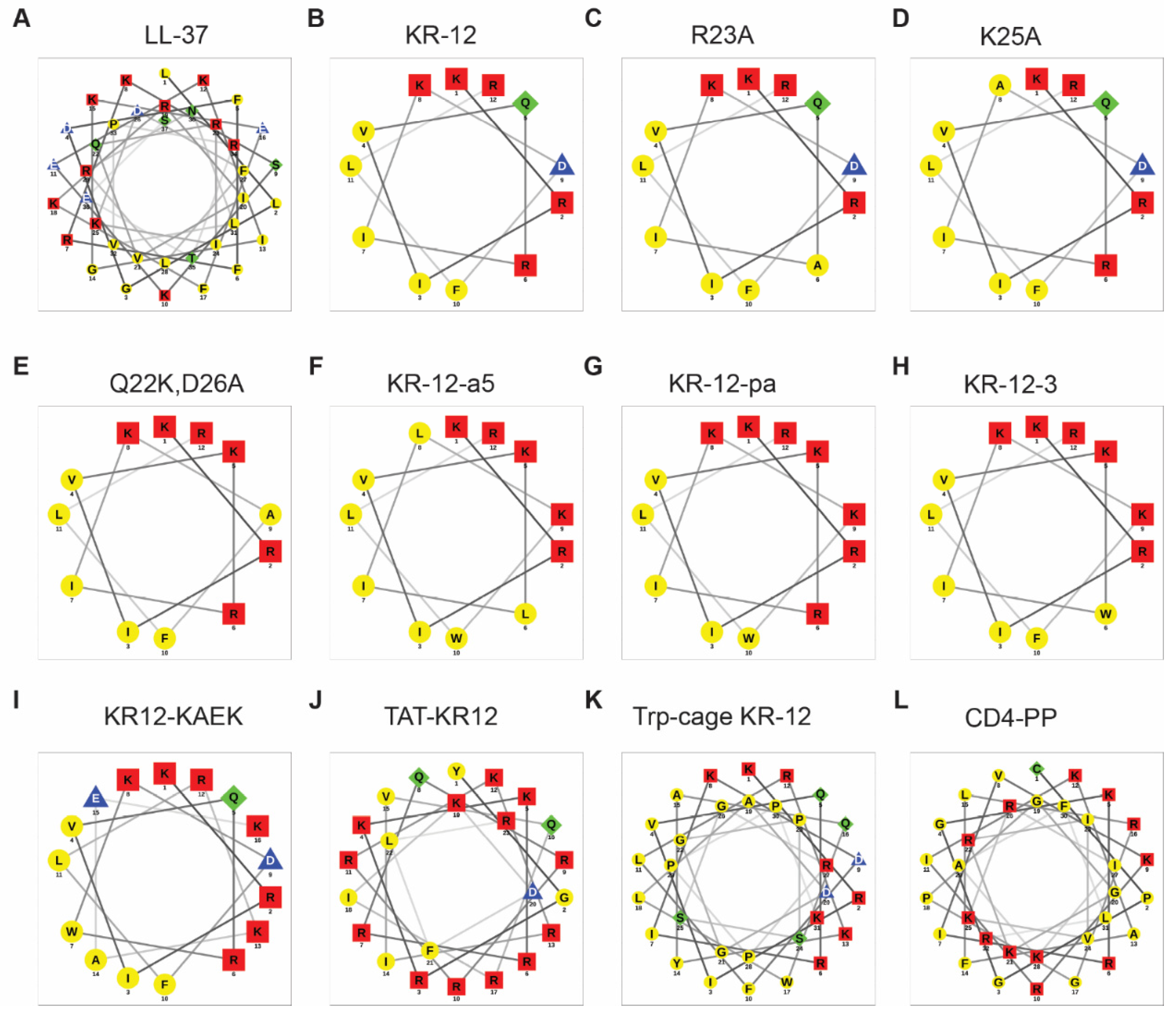
3.2. Alanine Scan
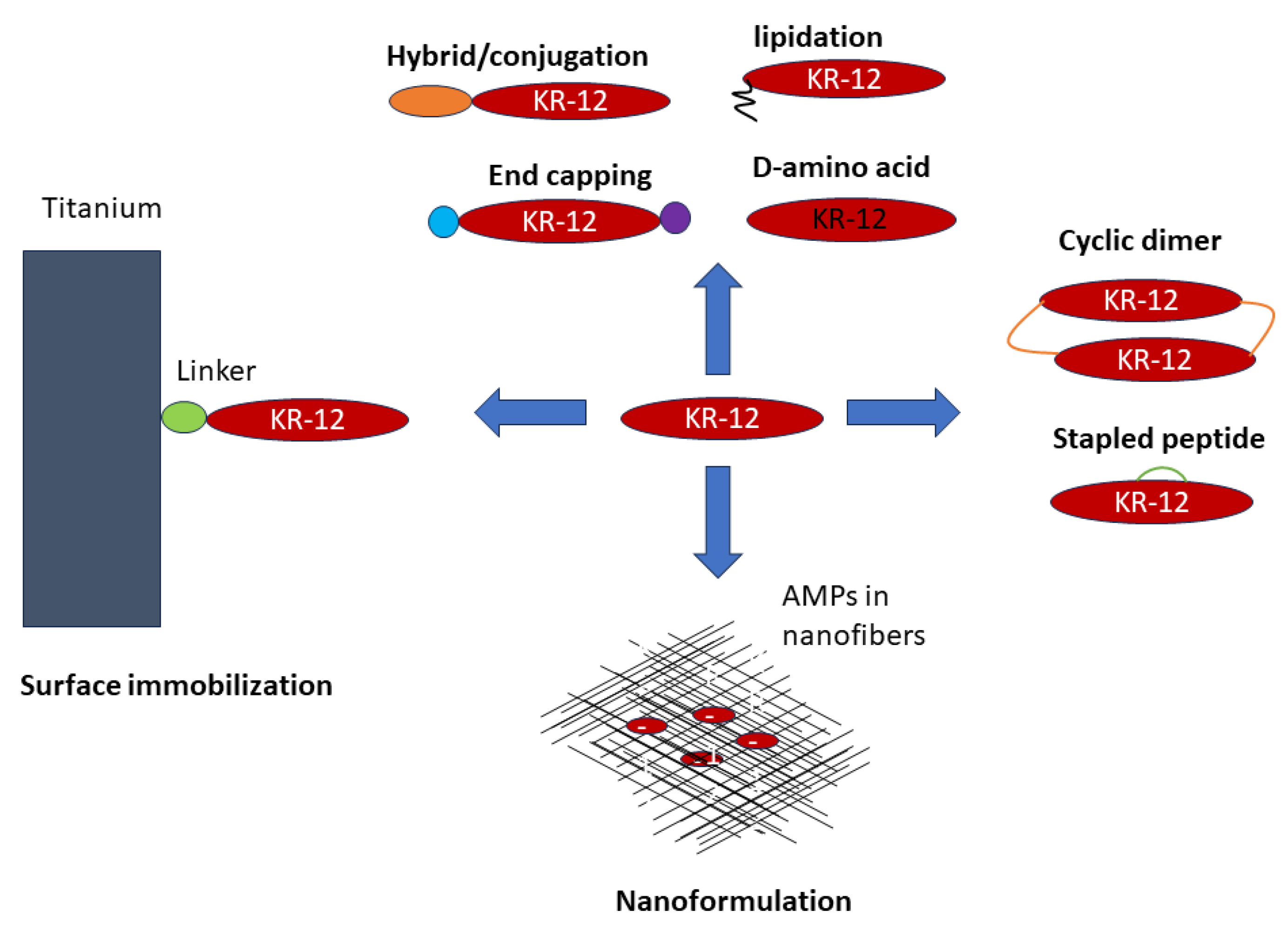
4. Innovative Engineering Strategies of KR-12
4.1. Linear Analogs
4.1.1. Amino Acid Substitutions
4.1.2. Peptide Hybrids and Conjugates with Enhanced Function
4.1.3. Lipopeptides: Conjugates with Organic Acids
4.2. Sidechain-Linked Peptides: Stapled KR-12
4.3. Head-to-Tail Cyclization of KR-12
4.4. Surface Immobilized KR-12
4.5. Peptide Formulation
5. Beyond KR-12: Discovery and Design of Even Shorter LL-37 Peptides
5.1. Short Lipopeptides Designed Based on KR-12 Segments
5.2. Design of LL-37mini Based on KR-8 Identified in Diluted Media
6. Safety and Efficacy of KR-12 Engineered Peptides in Animal Models
6.1. In Vivo Safety Evaluation
6.1.1. Systemic Toxicity
6.1.2. Topical Ototoxicity of KR-12-a2 to Guinea Pigs
6.2. In Vivo Efficacy Evaluation of KR-12 Peptides in Topical and Systemic Animal Models
6.2.1. Catheter Associated Bacterial Biofilm Mice Model
6.2.2. Wound Healing Models
6.2.3. LPS-Induced Bone Erosion in Mice Models
6.2.4. Evaluation of Osteointegration of Surface Immobilized KR-12 in Rat Models
6.2.5. Induced Colitis in a Mouse Model
6.2.6. E. coli-Induced Lethal Sepsis in a Murine Model
6.2.7. Systemic Efficacy of Lipopeptide C10-KR8
7. Conclusions and Perspectives
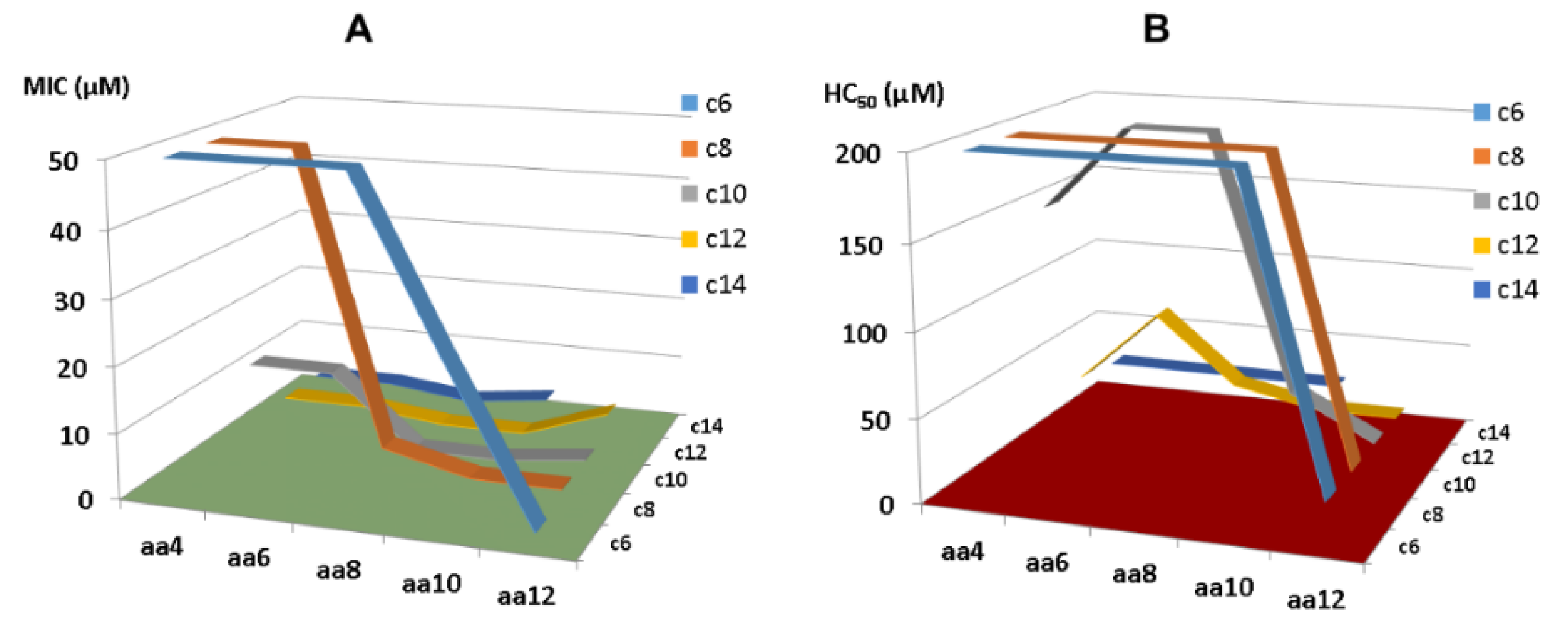
- As the smallest antibacterial peptide, the antibacterial activity of KR-12 is very susceptible to chemical modification. Even terminal capping has an evident effect on peptide activity. Our media dilution has deepened our understanding of the activity differences of these peptides reported from different labs [83]. To facilitate activity comparison from different laboratories, it is best to conduct antimicrobial assays by following the Clinical and Laboratory Standards Institute (CLSI) standard procedure. However, one may use a more sensitive media condition in initial peptide screening to identify more antimicrobial templates. Under such a condition, we identified KR-8 and RIK-10, two active peptides smaller than KR-12. These minimized LL-37 peptides enriched our antimicrobial reservoir.
- Peptide conjugation can enrich the structure and function of KR-12, including cell penetration, carbohydrate binding, and structural stabilization [48,50,54]. KR-12 can also be conjugated with fatty acids, providing another route to selective and potent peptides shorter than KR-12. As a special conjugation, KR-12 has been covalently immobilized via different chemistries onto biomaterials to confer antimicrobial and anti-inflammatory activities. In the presence of a flexible linker, the activity spectrum of free KR-12 is maintained.
- The first common theme in KR-12 engineering is to enhance peptide activity [39,85]. For this purpose, both basic and hydrophobic amino acids are increased. The 3D structure of KR-12 in complex with D8PG [32] and SDS [115] laid a solid basis for peptide design. Q22 and D26 are most obvious and logical to change. Evidently, peptide toxicity sets a limit to these changes. The increase in peptide toxicity is more pronounced, however, when interfacial R23 and K25 of KR-12 are converted to a hydrophobic leucine [80]. It is an art to control peptide hydrophobicity to the right level so that the peptide construct retains potency and lacks toxicity [12,35]. Similarly, one cannot keep increasing basic lysine/arginine since toxicity also rises once a certain limit is passed [153].
- Another desired goal is to confer stability to KR-12 to increase its bioavailability. Both simple and complex methods have been utilized. Simple practices include C-terminal amidation, N-terminal acetylation, D-amino acid incorporation (partially or in full), and lipidation, whereas conjugation with a Trp-cage, creation of hydrophobic staples and head-to-tail cyclization are more complex engineering practices. Most of the more stable forms of KR-12 appeared after 2020 (see timeline in Table 1). While lipopeptides such as C10-KR8d gains stability by incorporating D-amino acids [51], sidechain-linked staples stabilize the helical structure of KR-12 to diminish protease digestion [55]. Inspired by natural cyclotides, the engineered cyclic KR-12 construct becomes more stable because of unexposed termini and structural stabilization [114,116]. One possible reason for the dimeric design is that direct head-to-tail cyclization of a short LL-37 fragment may not be feasible due to the steric strain in the cyclic structure. Formulation of KR-12 in polymers can also protect the peptide from degradation [130].
- Because LL-37 is known to be inactivated by human serum [118], a third important goal of KR-12 engineering is to increase peptide bioavailability. Conversion of L-amino acids of C10-KR8 to D-amino acids provides a classic but useful strategy. With a different chiral configuration, the association of C10-KR8d with proteases and other proteins is minimized [51]. The cyclic peptide also retained activity in serum [116]. In addition, multiple KR-12 constructs have shown in vivo efficacy in topical models (catheter or wound healing) (Table 2). These innovative KR-12 constructs have enriched our LL-37 based defense arsenal.
- Mechanistically, KR-12 with a minimal antibacterial sequence can cluster anionic lipids and permeabilize bacterial membranes similar to its parent peptide LL-37 [43,75,154]. Most of the enhanced KR-12 peptides discussed herein can also damage bacterial membranes [43,55,116]. Recently, attention has also been paid to peptide amyloid formation, which is proposed to play a role in bacterial killing. Although oligomers (α-amyloids?) have been detected for KR-12, FK-13/LL-37(17-29) [85,86], and LL-37, all helical peptides [65,89,90,91], further studies are required to determine to what extent such a process contributes to bacterial killing.
- We should not forget that bacteria can respond to LL-37 and its peptides in numerous ways, including membrane modification to diminish peptide killing [10]. For this purpose, bacterial transposon libraries and proteomic studies [155,156] provide powerful tools for mapping bacterial resistant genes. However, we should emphasize that LL-37 peptides, including the KR-12 constructs engineered and discussed herein, remain potent against drug-resistant pathogens, persisters, and their biofilms [51,52,121,122]. Bacteria did not develop resistance to KR-12-derived peptides such as C10-KR8d or LL-37mini in multiple passage experiments [51,83].
- Peptide cost and large-scale production: It is critical to address peptide synthesis, purification, and large-scale production challenges [158]. While small peptides such as C10-KR8 with only eight residues (Table 1) can take full advantage of the existing large-scale industrial peptide synthesis capability, longer constructs such as KR-12 conjugated with Trp-cage (Table 1) may be produced via fermentation. However, we may not be able to produce cyclic dimeric KR-12 entirely by fermentation until the last step of the head-to-tail cyclization can be accomplished enzymatically. The scalability is equally important for formulated or surface immobilization KR-12. In addition, one should avoid activity loss due to chemical modification during storage of a large quantity of highly purified peptide therapeutics. Some AMPs can lose activity due to oxidation (e.g., methionine) [159,160], while succinimide (intramolecular cyclization of the sidechain of aspartic acid with its backbone amide) KR-12 showed reduced activity, helicity, and serum stability [161].
- Safety: Toxicity studies of KR-12 constructs are very limited and more thorough evaluations are needed both in vitro (using different cell lines) and in vivo (two animal models, per U.S. Food and Drug Administration (FDA) regulation). This safety requirement need be extended to chemicals used for formulation. The use of FDA-approved formulants is a shortcut. The safety evaluation may include the potential impact of new drugs on microbiomes as well.
- Efficacy in animal models: Continued studies are required to evaluate the efficacy of various constructs of KR-12 in proper animal models that mimic different diseases. Rigorous preclinical studies are necessary to evaluate PK/pharmacodynamics (PD) and biodistribution of KR-12 constructs in animal models and, eventually, in humans. Age and sex differences to treatment outcomes should be considered. Similar studies on formulated KR-12 are also needed. Ultimately, one may also make use of other cell models or even newly developed AI algorithms for efficacy and safety evaluation without the use of animals [162].
- Application scope: Future studies may find practical applications for various engineered constructs of KR-12 described herein and to be engineered. Depending on the problem, a proper construct may be chosen. For the sake of cost, one would embrace the simplest molecular design. When KR-12 constructs alone do not produce the desired treatment outcomes, combination treatment may be considered [140,163,164]. In addition, formulations can expand their application scope. Natural vehicles such as exosome-loaded LL-37 have recently been reported to protect against Zika virus infection [165]. It is exciting that precise nanopores produced via 3D printing can also work synergistically with LL-37 mimicking peptides to promote wound healing [166]. These delivery vehicles may increase the therapeutic efficacy of AMPs and their inducers if sufficient attention can also be paid to safety.
- Regulatory and clinical pathways to novel peptide antibiotics: Understanding the regulatory requirements and clinical development pathways for KR-12 constructs in particular and antimicrobial peptides in general, including formulations and immobilization, will facilitate their translation from laboratory to clinical settings. Collaborative efforts among researchers, industry, and regulatory agencies are required to streamline the development, to establish proper standards for peptide drugs, and to provide minimal equipment for transporting and storing peptide drugs.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations: The Review on Antimicrobial Resistance; Wellcome Trust: London, UK, 2016. [Google Scholar]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Schweizer, H.P. Understanding Efflux in Gram-Negative Bacteria: Opportunities for Drug Discovery. Expert. Opin. Drug Discov. 2012, 7, 633–642. [Google Scholar] [CrossRef]
- Hommes, J.W.; Surewaard, B.G.J. Intracellular Habitation of Staphylococcus aureus: Molecular Mechanisms and Prospects for Antimicrobial Therapy. Biomedicines 2022, 10, 1804. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Alford, M.A.; Haney, E.F. Antibiofilm Activity of Host Defence Peptides: Complexity Provides Opportunities. Nat. Rev. Microbiol. 2021, 19, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Shai, Y. Temporins and Their Synergism against Gram-Negative Bacteria and in Lipopolysaccharide Detoxification. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial Host Defence Peptides: Functions and Clinical Potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Golla, R.M.; Mishra, B.; Dang, X.; Lakshmaiah Narayana, J.; Li, A.; Xu, L.; Wang, G. Resistome of Staphylococcus aureus in Response to Human Cathelicidin LL-37 and Its Engineered Antimicrobial Peptides. ACS Infect. Dis. 2020, 6, 1866–1881. [Google Scholar] [CrossRef]
- Wang, G. The Antimicrobial Peptide Database Is 20 Years Old: Recent Developments and Future Directions. Protein Sci. 2023, 32, e4778. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Human Antimicrobial Peptides and Proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef] [PubMed]
- Schutte, B.C.; Mitros, J.P.; Bartlett, J.A.; Walters, J.D.; Jia, H.P.; Welsh, M.J.; Casavant, T.L.; McCray, P.B. Discovery of Five Conserved β-Defensin Gene Clusters Using a Computational Search Strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 2129–2133. [Google Scholar] [CrossRef]
- Sørensen, O.; Arnljots, K.; Cowland, J.B.; Bainton, D.F.; Borregaard, N. The Human Antibacterial Cathelicidin, HCAP-18, Is Synthesized in Myelocytes and Metamyelocytes and Localized to Specific Granules in Neutrophils. Blood 1997, 9090, 2796–2803. [Google Scholar] [CrossRef]
- Cowland, J.B.; Johnsen, A.H.; Borregaard, N. HCAP-18, a Cathelin/pro-Bactenecin-like Protein of Human Neutrophil Specific Granules. FEBS Lett. 1995, 368, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Frohm, M.; Agerberth, B.; Ahangari, G.; Ståhle-Bäckdahl, M.; Lidén, S.; Wigzell, H.; Gudmundsson, G.H. The Expression of the Gene Coding for the Antibacterial Peptide LL-37 Is Induced in Human Keratinocytes during Inflammatory Disorders. J. Biol. Chem. 1997, 272, 15258–15263. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, O.E.; Gram, L.; Johnsen, A.H.; Andersson, E.; Bangsbøll, S.; Tjabringa, G.S.; Hiemstra, P.S.; Malm, J.; Egesten, A.; Borregaard, N. Processing of Seminal Plasma HCAP-18 to ALL-38 by Gastricsin. A Novel Mechanism of Generating Antimicrobial Peptides in Vagina. J. Biol. Chem. 2003, 278, 28540–28546. [Google Scholar] [CrossRef]
- Meister, M.; Lemaitre, B.; Hoffmann, J.A. Antimicrobial Peptide Defense in Drosophila. BioEssays 1997, 19, 1019–1026. [Google Scholar] [CrossRef]
- Rock, F.L.; Hardiman, G.; Timans, J.C.; Kastelein, R.A.; Bazan, J.F. A Family of Human Receptors Structurally Related to Drosophila Toll. Proc. Natl. Acad. Sci. USA 1998, 95, 588–593. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like Receptor Triggering of a Vitamin D-Mediated Human Antimicrobial Response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Chen, X.; Niyonsaba, F.; Ushio, H.; Okuda, D.; Nagaoka, I.; Ikeda, S.; Okumura, K.; Ogawa, H. Synergistic effect of antibacterial agents human beta-defensins, cathelicidin LL-37 and lysozyme against Staphylococcus aureus and Escherichia coli. J. Dermatol. Sci. 2005, 40, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, P.; Scragg, R. A Short History of Phototherapy, Vitamin D and Skin Disease. Photochem. Photobiol. Sci. 2017, 16, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Dürr, U.H.N.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the Only Human Member of the Cathelicidin Family of Antimicrobial Peptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1408–1425. [Google Scholar] [CrossRef]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A Comprehensive Summary of LL-37, the Factoctum Human Cathelicidin Peptide. Cell Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef]
- Wang, G.; Narayana, J.L.; Mishra, B.; Zhang, Y.; Wang, F.; Wang, C.; Zarena, D.; Lushnikova, T.; Wang, X. Design of Antimicrobial Peptides: Progress Made with Human Cathelicidin LL-37. Adv. Exp. Med. Biol. 2019, 1117, 215–240. [Google Scholar] [CrossRef]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human Host Defense Peptide LL-37 Prevents Bacterial Biofilm Formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.E.; Davidson, D.J.; Hancock, R.E.W. Immunomodulatory Properties of Defensins and Cathelicidins. Curr. Top. Microbiol. Immunol. 2006, 306, 27–66. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjya, S.; Zhang, Z.; Ramamoorthy, A. LL-37: Structures, Antimicrobial Activity, and Influence on Amyloid-Related Diseases. Biomolecules 2024, 14, 320. [Google Scholar] [CrossRef]
- Zanetti, M. Cathelicidins, Multifunctional Peptides of the Innate Immunity. J. Leukoc. Biol. 2004, 75, 39–48. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from Human Skin Commensal Bacteria Protect against Staphylococcus Aureus and Are Deficient in Atopic Dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef]
- Wang, G. Structures of Human Host Defense Cathelicidin LL-37 and Its Smallest Antimicrobial Peptide KR-12 in Lipid Micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef]
- Wang, T.-T.; Nestel, F.P.; Bourdeau, V.; Nagai, Y.; Wang, Q.; Liao, J.; Tavera-Mendoza, L.; Lin, R.; Hanrahan, J.W.; Mader, S.; et al. Cutting Edge: 1,25-Dihydroxyvitamin D3 Is a Direct Inducer of Antimicrobial Peptide Gene Expression. J. Immunol. 2004, 173, 2909–2912. [Google Scholar] [CrossRef] [PubMed]
- Missailidis, C.; Sørensen, N.; Ashenafi, S.; Amogne, W.; Kassa, E.; Bekele, A.; Getachew, M.; Gebreselassie, N.; Aseffa, A.; Aderaye, G.; et al. Vitamin D and Phenylbutyrate Supplementation Does Not Modulate Gut Derived Immune Activation in HIV-1. Nutrients 2019, 11, 1675. [Google Scholar] [CrossRef]
- Miranda, E.; Bramono, K.; Yunir, E.; Reksodiputro, M.H.; Suwarsa, O.; Rengganis, I.; Harahap, A.R.; Subekti, D.; Suwarto, S.; Hayun, H.; et al. Efficacy of LL-37 Cream in Enhancing Healing of Diabetic Foot Ulcer: A Randomized Double-Blind Controlled Trial. Arch. Dermatol. Res. 2023, 315, 2623–2633. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Kiattiburut, W.; Khongkha, T.; Burke Schinkel, S.C.; Lunn, Y.; Decker, A.P.; Mohammadi, A.; Vera-Cruz, A.; Misra, A.; Angel, J.B.; et al. 17BIPHE2, an Engineered Cathelicidin Antimicrobial Peptide with Low Susceptibility to Proteases, Is an Effective Spermicide and Microbicide against Neisseria Gonorrhoeae. Hum. Reprod. 2022, 37, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.C.; Lee, A.H.-Y.; Hancock, R.E.W. Mechanisms of the Innate Defense Regulator Peptide-1002 Anti-Inflammatory Activity in a Sterile Inflammation Mouse Model. J. Immunol. 2017, 199, 3592–3603. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.K.; Wang, G.; Coffelt, S.B.; Betancourt, A.M.; Lee, C.W.; Fan, D.; Wu, K.; Yu, J.; Sung, J.J.Y.; Cho, C.H. Emerging Roles of the Host Defense Peptide LL-37 in Human Cancer and Its Potential Therapeutic Applications. Int. J. Cancer 2010, 127, 1741–1747. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Han, H.; Miller, D.W.; Wang, G. Solution Structures of Human Ll-37 Fragments and NMR-Based Identification of a Minimal Membrane-Targeting Antimicrobial and Anticancer Region. J. Am. Chem. Soc. 2006, 128, 5776–5785. [Google Scholar] [CrossRef]
- Ren, S.X.; Shen, J.; Cheng, A.S.L.; Lu, L.; Chan, R.L.Y.; Li, Z.J.; Wang, X.J.; Wong, C.C.M.; Zhang, L.; Ng, S.S.M.; et al. FK-16 Derived from the Anticancer Peptide LL-37 Induces Caspase-Independent Apoptosis and Autophagic Cell Death in Colon Cancer Cells. PLoS ONE 2013, 8, e63641. [Google Scholar] [CrossRef]
- Lu, F.; Zhu, Y.; Zhang, G.; Liu, Z. Renovation as Innovation: Repurposing Human Antibacterial Peptide LL-37 for Cancer Therapy. Front. Pharmacol. 2022, 13, 944147. [Google Scholar] [CrossRef]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wójcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J.; et al. Degradation of Human Antimicrobial Peptide LL-37 by Staphylococcus Aureus-Derived Proteinases. Antimicrob. Agents Chemother. 2004, 48, 4673–4679. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Epand, R.F.; Epand, R.M.; Wang, G. Structural Location Determines Functional Roles of the Basic Amino Acids of KR-12, the Smallest Antimicrobial Peptide from Human Cathelicidin LL-37. RSC Adv. 2013, 3, 19560–19571. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Min, H.J.; Lee, C.W. NMR Structure and Bactericidal Activity of KR-12 Analog Derived from Human LL-37 as a Potential Cosmetic Preservative. J. Anal. Sci. Technol. 2020, 11, 14. [Google Scholar] [CrossRef]
- Nie, B.; Ao, H.; Chen, C.; Xie, K.; Zhou, J.; Long, T.; Tang, T.; Yue, B. Covalent Immobilization of KR-12 Peptide onto a Titanium Surface for Decreasing Infection and Promoting Osteogenic Differentiation. RSC Adv. 2016, 6, 46733–46743. [Google Scholar] [CrossRef]
- Song, D.W.; Kim, S.H.; Kim, H.H.; Lee, K.H.; Ki, C.S.; Park, Y.H. Multi-Biofunction of Antimicrobial Peptide-Immobilized Silk Fibroin Nanofiber Membrane: Implications for Wound Healing. Acta Biomater. 2016, 39, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-Derived Short Antimicrobial Peptide KR-12-A5 and Its D-Amino Acid Substituted Analogs with Cell Selectivity, Anti-Biofilm Activity, Synergistic Effect with Conventional Antibiotics, and Anti-Inflammatory Activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Chen, C.; Lyu, Z.; Zhang, S.; Wang, Y.; Nie, B.; Yue, B. Overcoming Planktonic and Intracellular Staphylococcus Aureus-Associated Infection with a Cell-Penetrating Peptide-Conjugated Antimicrobial Peptide. ACS Infect. Dis. 2020, 6, 3147–3162. [Google Scholar] [CrossRef] [PubMed]
- Kamysz, E.; Sikorska, E.; Jaśkiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Lipidated Analogs of the Ll-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int. J. Mol. Sci. 2020, 21, 887. [Google Scholar] [CrossRef]
- Preußke, N.; Lipfert, M.; Rothemund, S.; Leippe, M.; Sönnichsen, F.D. Designed Trp-Cage Proteins with Antimicrobial Activity and Enhanced Stability. Biochemistry 2021, 60, 3187–3199. [Google Scholar] [CrossRef]
- Lakshmaiah Narayana, J.; Golla, R.; Mishra, B.; Wang, X.; Lushnikova, T.; Zhang, Y.; Verma, A.; Kumar, V.; Xie, J.; Wang, G. Short and Robust Anti-Infective Lipopeptides Engineered Based on the Minimal Antimicrobial Peptide KR12 of Human LL-37. ACS Infect. Dis. 2021, 7, 1795–1808. [Google Scholar] [CrossRef]
- White, J.K.; Muhammad, T.; Alsheim, E.; Mohanty, S.; Blasi-Romero, A.; Gunasekera, S.; Strömstedt, A.A.; Ferraz, N.; Göransson, U.; Brauner, A. A Stable Cyclized Antimicrobial Peptide Derived from LL-37 with Host Immunomodulatory Effects and Activity against Uropathogens. Cell. Mol. Life Sci. 2022, 79, 411. [Google Scholar] [CrossRef]
- Ki, M.R.; Kim, S.H.; Park, T.I.; Pack, S.P. Self-Entrapment of Antimicrobial Peptides in Silica Particles for Stable and Effective Antimicrobial Peptide Delivery System. Int. J. Mol. Sci. 2023, 24, 16423. [Google Scholar] [CrossRef] [PubMed]
- van Zyl, E.M.; Coburn, J.M. Functionalization of Bacterial Cellulose with the Antimicrobial Peptide KR-12 via Chimerical Cellulose-Binding Peptides. Int. J. Mol. Sci. 2024, 25, 1462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, M.; Wang, Z.; Liu, Z.; Chen, S.; Li, X.; Shi, Y.; Hu, H. Discovery of Novel Antibacterial Agent for the Infected Wound Treatment: All-Hydrocarbon Stapling Optimization of LL-37. Theranostics 2024, 14, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Nell, M.J.; Tjabringa, G.S.; Wafelman, A.R.; Verrijk, R.; Hiemstra, P.S.; Drijfhout, J.W.; Grote, J.J. Development of Novel LL-37 Derived Antimicrobial Peptides with LPS and LTA Neutralizing and Antimicrobial Activities for Therapeutic Application. Peptides 2006, 27, 649–660. [Google Scholar] [CrossRef]
- Nagant, C.; Pitts, B.; Nazmi, K.; Vandenbranden, M.; Bolscher, J.G.; Stewart, P.S.; Dehaye, J.P. Identification of Peptides Derived from the Human Antimicrobial Peptide LL-37 Active against Biofilms Formed by Pseudomonas Aeruginosa Using a Library of Truncated Fragments. Antimicrob. Agents Chemother. 2012, 56, 5698–5708. [Google Scholar] [CrossRef]
- Braff, M.H.; Hawkins, M.A.; Di Nardo, A.; Lopez-Garcia, B.; Howell, M.D.; Wong, C.; Lin, K.; Streib, J.E.; Dorschner, R.; Leung, D.Y.M.; et al. Structure-Function Relationships among Human Cathelicidin Peptides: Dissociation of Antimicrobial Properties from Host Immunostimulatory Activities. J. Immunol. 2005, 174, 4271–4278. [Google Scholar] [CrossRef]
- De Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; De Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; Van Der Heijde, T.; Boekema, B.K.; et al. The Antimicrobial Peptide SAAP-148 Combats Drug-Resistant Bacteria and Biofilms. Sci. Transl. Med. 2018, 10, eaan4044. [Google Scholar] [CrossRef]
- Wang, G.; Epand, R.F.; Mishra, B.; Lushnikova, T.; Thomas, V.C.; Bayles, K.W.; Epand, R.M. Decoding the Functional Roles of Cationic Side Chains of the Major Antimicrobial Region of Human Cathelicidin LL-37. Antimicrob. Agents Chemother. 2012, 56, 845–856. [Google Scholar] [CrossRef]
- He, M.; Zhang, H.; Li, Y.; Wang, G.; Tang, B.; Zhao, J.; Huang, Y.; Zheng, J. Cathelicidin-Derived Antimicrobial Peptides Inhibit Zika Virus Through Direct Inactivation and Interferon Pathway. Front Immunol. 2018, 9, 722. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Peterkofsky, A.; Wang, G. NMR Studies of Aurein 1.2 Analogs. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Saporito, P.; Vang Mouritzen, M.; Løbner-Olesen, A.; Jenssen, H. LL-37 Fragments Have Antimicrobial Activity against Staphylococcus Epidermidis Biofilms and Wound Healing Potential in HaCaT Cell Line. J. Pept. Sci. 2018, 24, e3080. [Google Scholar] [CrossRef] [PubMed]
- Chanci, K.; Diosa, J.; Giraldo, M.A.; Mesa, M. Physical Behavior of KR-12 Peptide on Solid Surfaces and Langmuir-Blodgett Lipid Films: Complementary Approaches to Its Antimicrobial Mode against S. aureus. Biochim. Biophys. Acta Biomembr. 2022, 1864, 183779. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and Organization of the Human Antimicrobial Peptide LL-37 in Phospholipid Membranes: Relevance to the Molecular Basis for Its Non-Cell-Selective Activity. Biochem. J. 1999, 341 Pt 3, 501–513. [Google Scholar] [CrossRef]
- Cutrona, K.J.; Kaufman, B.A.; Figueroa, D.M.; Elmore, D.E. Role of Arginine and Lysine in the Antimicrobial Mechanism of Histone-Derived Antimicrobial Peptides. FEBS Lett. 2015, 589 Pt B, 3915–3920. [Google Scholar] [CrossRef]
- Amirkhanov, N.V.; Bardasheva, A.V.; Tikunova, N.V.; Pyshnyi, D.V. Synthetic Antimicrobial Peptides: III—Effect of Cationic Groups of Lysine, Arginine, and Histidine on Antimicrobial Activity of Peptides with a Linear Type of Amphipathicity. Russ. J. Bioorg. Chem. 2021, 47, 681–690. [Google Scholar] [CrossRef]
- Arias, M.; Piga, K.B.; Hyndman, M.E.; Vogel, H.J. Improving the Activity of Trp-Rich Antimicrobial Peptides by Arg/Lys Substitutions and Changing the Length of Cationic Residues. Biomolecules 2018, 8, 19. [Google Scholar] [CrossRef]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR Drug Discovery: Integrating Solution NMR Data with Crystal and Cryo-EM Structures. Nat. Rev. Drug Discov. 2018, 18, 59–82. [Google Scholar] [CrossRef]
- Cushley, R.J.; Okon, M. NMR Studies of Lipoprotein Structure. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 177–206. [Google Scholar] [CrossRef]
- Schibli, D.J.; Hwang, P.M.; Vogel, H.J. The Structure of the Antimicrobial Active Center of Lactoferricin B Bound to Sodium Dodecyl Sulfate Micelles. FEBS Lett. 1999, 446, 213–217. [Google Scholar] [CrossRef]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E.W. Structure of the Bovine Antimicrobial Peptide Indolicidin Bound to Dodecylphosphocholine and Sodium Dodecyl Sulfate Micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef] [PubMed]
- Keifer, P.A.; Peterkofsky, A.; Wang, G. Effects of Detergent Alkyl Chain Length and Chemical Structure on the Properties of a Micelle-Bound Bacterial Membrane Targeting Peptide. Anal. Biochem. 2004, 331, 33–39. [Google Scholar] [CrossRef]
- Wang, G. Determination of Solution Structure and Lipid Micelle Location of an Engineered Membrane Peptide by Using One NMR Experiment and One Sample. Biochim. Biophys. Acta Biomembr. 2007, 1768, 3271–3281. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Wang, G.; Berno, B.; Epand, R.M. Lipid Segregation Explains Selective Toxicity of a Series of Fragments Derived from the Human Cathelicidin LL-37. Antimicrob. Agents Chemother. 2009, 53, 3705–3714. [Google Scholar] [CrossRef] [PubMed]
- Mól, A.R.; Castro, M.S.; Wagner, F. NetWheels: A Web Application to Create High Quality Peptide Helical Wheel and Net Projections. J. Bioinform. Syst. Biol. 2024, 7, 98–100. [Google Scholar] [CrossRef]
- Lee, I.H.; Zhao, C.; Nguyen, T.; Menzel, L.; Waring, A.J.; Sherman, M.A.; Lehrer, R.I. Clavaspirin, an antibacterial and haemolytic peptide from Styela clava. J. Pept. Res. 2001, 58, 445–456. [Google Scholar] [CrossRef]
- Jung, D.; Rozek, A.; Okon, M.; Hancock, R.E.W. Structural Transitions as Determinants of the Action of the Calcium-Dependent Antibiotic Daptomycin. Chem. Biol. 2004, 11, 949–957. [Google Scholar] [CrossRef]
- Brogden, K.A.; De Lucca, A.J.; Bland, J.; Elliott, S. Isolation of an Ovine Pulmonary Surfactant-Associated Anionic Peptide Bactericidal for Pasteurella Haemolytica. Proc. Natl. Acad. Sci. USA 1996, 93, 412–416. [Google Scholar] [CrossRef]
- Jacob, B.; Park, I.S.; Bang, J.K.; Shin, S.Y. Short KR-12 Analogs Designed from Human Cathelicidin LL-37 Possessing Both Antimicrobial and Antiendotoxic Activities without Mammalian Cell Toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef]
- Luo, Y.; McLean, D.T.F.; Linden, G.J.; McAuley, D.F.; McMullan, R.; Lundy, F.T. The Naturally Occurring Host Defense Peptide, LL-37, and Its Truncated Mimetics KE-18 and KR-12 Have Selected Biocidal and Antibiofilm Activities against Candida albicans, Staphylococcus aureus, and Escherichia coli In Vitro. Front. Microbiol. 2017, 8, 544. [Google Scholar] [CrossRef]
- Turner, J.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I. Activities of LL-37, a Cathelin-Associated Antimicrobial Peptide of Human Neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. [Google Scholar] [CrossRef]
- Mechesso, A.F.; Su, Y.; Xie, J.; Wang, G. Enhanced Antimicrobial Screening Sensitivity Enabled the Identification of an Ultrashort Peptide KR-8 for Engineering of LL-37mini to Combat Drug-Resistant Pathogens. ACS Infect. Dis. 2023, 9, 2215–2225. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (MIC) of Antimicrobial Substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Henning-Knechtel, A.; Österlund, N.; Wu, J.; Wang, G.; Gräslund, R.A.O.; Kirmizialtin, S.; Luo, J. Oligomer Dynamics of LL-37 Truncated Fragments Probed by α-Hemolysin Pore and Molecular Simulations. Small 2023, 19, e2206232. [Google Scholar] [CrossRef] [PubMed]
- Engelberg, Y.; Landau, M. The Human LL-37(17-29) Antimicrobial Peptide Reveals a Functional Supramolecular Structure. Nat. Commun. 2020, 11, 3894. [Google Scholar] [CrossRef] [PubMed]
- Bücker, R.; Seuring, C.; Cazey, C.; Veith, K.; García-Alai, M.; Grünewald, K.; Landau, M. The Cryo-EM Structures of Two Amphibian Antimicrobial Cross-β Amyloid Fibrils. Nat. Commun. 2022, 13, 4356. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, M.; Tsuji, R.; Suenaga, S.; Matsuo, K. Formation of β-Strand Oligomers of Antimicrobial Peptide Magainin 2 Contributes to Disruption of Phospholipid Membrane. Membranes 2022, 12, 131. [Google Scholar] [CrossRef]
- Xhindoli, D.; Pacor, S.; Guida, F.; Antcheva, N.; Tossi, A. Native Oligomerization Determines the Mode of Action and Biological Activities of Human Cathelicidin LL-37. Biochem. J. 2014, 457, 263–275. [Google Scholar] [CrossRef]
- Sancho-Vaello, E.; Gil-Carton, D.; François, P.; Bonetti, E.J.; Kreir, M.; Pothula, K.R.; Kleinekathöfer, U.; Zeth, K. The Structure of the Antimicrobial Human Cathelicidin LL-37 Shows Oligomerization and Channel Formation in the Presence of Membrane Mimics. Sci. Rep. 2020, 10, 17356. [Google Scholar] [CrossRef]
- Zhang, Y.; Narayana, J.L.; Wu, Q.; Dang, X.; Wang, G. Structure and Activity of a Selective Antibiofilm Peptide Sk-24 Derived from the Nmr Structure of Human Cathelicidin Ll-37. Pharmaceuticals 2021, 14, 1245. [Google Scholar] [CrossRef]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-Quality 3D Structures Shine Light on Antibacterial, Anti-Biofilm and Antiviral Activities of Human Cathelicidin LL-37 and Its Fragments. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2160–2172. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Alanine and Lysine Scans of the LL-37-Derived Peptide Fragment KR-12 Reveal Key Residues for Antimicrobial Activity. ChemBioChem 2018, 19, 931–939. [Google Scholar] [CrossRef]
- Wang, G. (Ed.) Antimicrobial Peptides: Discovery, Design, and Novel Therapeutic Strategies; CABI: Wallingford, UK, 2010. [Google Scholar]
- Wang, G. Unifying the Classification of Antimicrobial Peptides in the Antimicrobial Peptide Database. Methods Enzymol. 2022, 663, 1–18. [Google Scholar] [PubMed]
- Caiaffa, K.S.; dos Santos, V.R.; Abuna, G.F.; Santos-Filho, N.A.; Cilli, E.M.; Sakai, V.T.; Cintra, L.T.A.; Duque, C. Cytocompatibility and Synergy of EGCG and Cationic Peptides Against Bacteria Related to Endodontic Infections, in Planktonic and Biofilm Conditions. Probiotics Antimicrob. Proteins 2021, 13, 1808–1819. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, H.; Zhang, X.; Li, M.; Zhang, Q.; Wang, Y. Antibacterial and Anti-Inflammatory Properties of a Novel Antimicrobial Peptide Derived from LL-37. Antibiotics 2022, 11, 754. [Google Scholar] [CrossRef] [PubMed]
- da Silva, B.R.; Conrado, A.J.S.; Pereira, A.L.; Evaristo, F.F.V.; Arruda, F.V.S.; Vasconcelos, M.A.; Lorenzón, E.N.; Cilli, E.M.; Teixeira, E.H. Antibacterial Activity of a Novel Antimicrobial Peptide [W7]KR12-KAEK Derived from KR-12 against Streptococcus Mutans Planktonic Cells and Biofilms. Biofouling 2017, 33, 835–846. [Google Scholar] [CrossRef]
- Yang, H.; Lu, B.; Zhou, D.; Zhao, L.; Song, W.; Wang, L. Identification of the First Cathelicidin Gene from Skin of Chinese Giant Salamanders Andrias Davidianus with Its Potent Antimicrobial Activity. Dev. Comp. Immunol. 2017, 77, 141–149. [Google Scholar] [CrossRef]
- Ajish, C.; Yang, S.; Kumar, S.D.; Kim, E.Y.; Min, H.J.; Lee, C.W.; Shin, S.H.; Shin, S.Y. A Novel Hybrid Peptide Composed of LfcinB6 and KR-12-A4 with Enhanced Antimicrobial, Anti-Inflammatory and Anti-Biofilm Activities. Sci. Rep. 2022, 12, 4365. [Google Scholar] [CrossRef]
- Chu, C.H.; Mei, M.L.; Wu, W.K.K. Novel Dentotropic Antimicrobial Peptide to Prevent Dental Caries: Abridged Secondary Publication. Hong Kong Med. J. 2023, 29, 34–38. [Google Scholar]
- Lei, R.; Yang, C.; Sun, Y.; Li, D.; Hao, L.; Li, Y.; Wu, S.; Li, H.; Lan, C.; Fang, X. Turning Cationic Antimicrobial Peptide KR-12 into Self-Assembled Nanobiotics with Potent Bacterial Killing and LPS Neutralizing Activities. Nanoscale 2023, 16, 887–902. [Google Scholar] [CrossRef]
- Verdine, G.L.; Hilinski, G.J. Stapled Peptides for Intracellular Drug Targets. Methods Enzymol. 2012, 503, 3–33. [Google Scholar]
- Bird, G.H.; Madani, N.; Perry, A.F.; Princiotto, A.M.; Supko, J.G.; He, X.; Gavathiotis, E.; Sodroski, J.G.; Walensky, L.D. Hydrocarbon Double-Stapling Remedies the Proteolytic Instability of a Lengthy Peptide Therapeutic. Proc. Natl. Acad. Sci. USA 2010, 107, 14093–14098. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.L.; Cho, O.; Scanlon, D.B.; Booker, G.W.; Abell, A.D.; Wegener, K.L. The Key Position: Influence of Staple Location on Constrained Peptide Conformation and Binding. Org. Biomol. Chem. 2016, 14, 9731–9735. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing Theα-Helical Structural Stability of Stapled P53 Peptides: Molecular Dynamics Simulations and Analysis: Research Article. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of Stapled Antimicrobial Peptides That Are Stable, Nontoxic and Kill Antibiotic-Resistant Bacteria in Mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, R.; Chen, S.; Zou, Y.; Yan, L.; Zhao, L.; Li, X. Design, Synthesis and Antifungal Activity of Stapled Aurein1.2 Peptides. Antibiotics 2021, 1010, 956. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, M.; Macyszyn, J.; Miszkiewicz, J.; Grzela, R.; Trylska, J. Stapled Anoplin as an Antibacterial Agent. Front. Microbiol. 2021, 12, 772038. [Google Scholar] [CrossRef]
- You, Y.H.; Liu, H.Y.; Zhu, Y.Z.; Zheng, H. Rational Design of Stapled Antimicrobial Peptides. Amino Acids 2023, 55, 421–442. [Google Scholar] [CrossRef]
- Craik, D.J.; Čemažar, M.; Wang, C.K.L.; Daly, N.L. The Cyclotide Family of Circular Miniproteins: Nature’s Combinatorial Peptide Template. Biopolym.—Pept. Sci. Sect. 2006, 84, 250–266. [Google Scholar] [CrossRef]
- Tongaonkar, P.; Trinh, K.K.; Ouellette, A.J.; Selsted, M.E. Inhibition of MiR-146a Expression and Regulation of Endotoxin Tolerance by Rhesus Theta-Defensin-1. Mediat. Inflamm. 2023, 2023, 8387330. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Backbone Cyclization and Dimerization of LL-37-Derived Peptides Enhance Antimicrobial Activity and Proteolytic Stability. Front. Microbiol. 2020, 11, 168. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Strömstedt, A.A.; Gunasekera, S.; Göransson, U. Transforming Cross-Linked Cyclic Dimers of KR-12 into Stable and Potent Antimicrobial Drug Leads. Biomedicines 2023, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Sol, A.; Wang, G.; Blotnick, E.; Golla, R.; Bachrach, G.; Muhlrad, A. Interaction of the Core Fragments of the LL-37 Host Defense Peptide with Actin. RSC Adv. 2015, 5, 9361–9367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Agerberth, B.; Löthgren, A.; Almstedt, A.; Johansson, J. Apolipoprotein A-I Binds and Inhibits the Human Antibacterial/Cytotoxic Peptide LL-37. J. Biol. Chem. 1998, 273, 33115–33118. [Google Scholar] [CrossRef]
- Habash, M.; Reid, G. Microbial Biofilms: Their Development and Significance for Medical Device-Related Infections. J. Clin. Pharmacol. 1999, 39, 887–898. [Google Scholar] [CrossRef]
- Tram, M.K.; Schammel, J.; Vancavage, R.; Welliver, C.; Inouye, B.M. Emerging Strategies for the Prevention of Bacterial Biofilm in Prosthetic Surgery. Transl. Androl. Urol. 2024, 13, 833–845. [Google Scholar] [CrossRef]
- Wang, G.; Hanke, M.L.; Mishra, B.; Lushnikova, T.; Heim, C.E.; Thomas, V.C.; Bayles, K.W.; Kielian, T. Transformation of Human Cathelicidin LL-37 into Selective, Stable, and Potent Antimicrobial Compounds. ACS Chem. Biol. 2014, 9, 1997–2002. [Google Scholar] [CrossRef]
- Scheper, H.; Wubbolts, J.M.; Verhagen, J.A.M.; de Visser, A.W.; van der Wal, R.J.P.; Visser, L.G.; de Boer, M.G.J.; Nibbering, P.H. SAAP-148 Eradicates MRSA Persisters Within Mature Biofilm Models Simulating Prosthetic Joint Infection. Front. Microbiol. 2021, 12, 625952. [Google Scholar] [CrossRef]
- Gabriel, M.; Nazmi, K.; Veerman, E.C.; Amerongen, A.V.N.; Zentner, A. Preparation of LL-37-Grafted Titanium Surfaces with Bactericidal Activity. Bioconjug Chem. 2006, 17, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Titanium Surfaces Immobilized with the Major Antimicrobial Fragment FK-16 of Human Cathelicidin LL-37 Are Potent against Multiple Antibiotic-Resistant Bacteria. Biofouling 2017, 33, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, Y.; Masuda, T.; Isu, N.; Takai, M. Antimicrobial Peptide Assembly on Zwitterionic Polymer Films to Slow Down Biofilm Formation. Langmuir 2024, 40, 7029–7037. [Google Scholar] [CrossRef]
- Janczura, M.; Sip, S.; Cielecka-Piontek, J. The Development of Innovative Dosage Forms of the Fixed-Dose Combination of Active Pharmaceutical Ingredients. Pharmaceutics 2022, 14, 834. [Google Scholar] [CrossRef] [PubMed]
- Thimmiah, B.R.; Chien, B.T.C.; Fui, K.S.; Yon, L.S.; Nallathambi, G.; Jeevanandam, J.; Danquah, M.K. Nanoformulation of Peptides for Pharmaceutical Applications: In Vitro and In Vivo Perspectives. Appl. Sci. 2022, 1212, 12777. [Google Scholar] [CrossRef]
- Nugrahadi, P.P.; Hinrichs, W.L.J.; Frijlink, H.W.; Schöneich, C.; Avanti, C. Designing Formulation Strategies for Enhanced Stability of Therapeutic Peptides in Aqueous Solutions: A Review. Pharmaceutics 2023, 15, 935. [Google Scholar] [CrossRef]
- Su, Y.; Wang, H.; Mishra, B.; Lakshmaiah Narayana, J.; Jiang, J.; Reilly, D.A.; Hollins, R.R.; Carlson, M.A.; Wang, G.; Xie, J. Nanofiber Dressings Topically Delivering Molecularly Engineered Human Cathelicidin Peptides for the Treatment of Biofilms in Chronic Wounds. Mol. Pharm. 2019, 16, 2011–2020. [Google Scholar] [CrossRef]
- Decker, A.P.; Su, Y.; Mishra, B.; Verma, A.; Lushnikova, T.; Xie, J.; Wang, G. Peptide Stability Is Important but Not a General Requirement for Antimicrobial and Antibiofilm Activity in Vitro and in Vivo. Mol. Pharm. 2023, 20, 738–749. [Google Scholar] [CrossRef]
- Blasi-Romero, A.; Ångström, M.; Franconetti, A.; Muhammad, T.; Jiménez-Barbero, J.; Göransson, U.; Palo-Nieto, C.; Ferraz, N. KR-12 Derivatives Endow Nanocellulose with Antibacterial and Anti-Inflammatory Properties: Role of Conjugation Chemistry. ACS Appl. Mater. Interfaces 2023, 15, 24186–24196. [Google Scholar] [CrossRef]
- Mishra, B.; Lushnikova, T.; Wang, G. Small Lipopeptides Possess Anti-Biofilm Capability Comparable to Daptomycin and Vancomycin. RSC Adv. 2015, 5, 59758–59769. [Google Scholar] [CrossRef]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and Arginine-Rich Antimicrobial Peptides: Structures and Mechanisms of Action. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The Preference of Tryptophan for Membrane Interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef]
- Wang, G.; Pierens, G.K.; Treleaven, W.D.; Sparrow, J.T.; Cushley, R.J. Conformations of Human Apolipoprotein E(263-286) and E(267-289) in Aqueous Solutions of Sodium Dodecyl Sulfate by CD and 1H NMR. Biochemistry 1996, 35, 10358–10366. [Google Scholar] [CrossRef]
- Mishra, B.; Lushnikova, T.; Golla, R.M.; Wang, X.; Wang, G. Design and Surface Immobilization of Short Anti-Biofilm Peptides. Acta Biomater. 2017, 49, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zietz, C.M.; Mudgapalli, A.; Wang, S.; Wang, Z. The Evolution of the Antimicrobial Peptide Database over 18 Years: Milestones and New Features. Protein Sci. 2022, 31, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Ledesma, M.G.; Rodríguez, M.C.; Alva-Murillo, N.; Avila, E.E. The Antimicrobial Peptides LL-37, KR-20, FK-13 and KR-12 Inhibit the Growth of a Sensitive and a Metronidazole-Resistant Strain of Trichomonas Vaginalis. Parasitol. Res. 2022, 121, 3503–3512. [Google Scholar] [CrossRef]
- Sung, C.M.; Kim, H.C.; Cho, Y.B.; Shin, S.Y.; Jang, C.H. Evaluating the Ototoxicity of an Anti-MRSA Peptide KR-12-A2. Braz. J. Otorhinolaryngol. 2018, 84, 441–447. [Google Scholar] [CrossRef]
- Liu, M.; Ding, R.; Li, Z.; Xu, N.; Gong, Y.; Huang, Y.; Jia, J.; Du, H.; Yu, Y.; Luo, G. Hyaluronidase-Responsive Bactericidal Cryogel for Promoting Healing of Infected Wounds: Inflammatory Attenuation, ROS Scavenging, and Immune Regulation. Adv. Sci. 2024, 11, e2306602. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Xu, C.; Cao, Y.; Liu, S.; Reis, R.L.; Kundu, S.C.; Yang, X.; Xiao, B.; Duan, L. Transparent Silk Fibroin Film-Facilitated Infected-Wound Healing through Antibacterial, Improved Fibroblast Adhesion and Immune Modulation. J. Mater. Chem. B 2023, 12, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, S.; Nie, B.; Long, T.; Qu, X.; Yue, B. KR-12-A5 Reverses Adverse Effects of Lipopolysaccharides on HBMSC Osteogenic Differentiation by Influencing BMP/Smad and P38 MAPK Signaling Pathways. Front. Pharmacol. 2019, 10, 639. [Google Scholar] [CrossRef]
- Caiaffa, K.S.; Massunari, L.; Danelon, M.; Abuna, G.F.; Bedran, T.B.L.; Santos-Filho, N.A.; Spolidorio, D.M.P.; Vizoto, N.L.; Cilli, E.M.; Duque, C. KR-12-a5 is a non-cytotoxic agent with potent antimicrobial effects against oral pathogens. Biofouling 2017, 33, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, J.; Chen, J.; Nie, B.; Yue, B.; Zhang, W.; Lyu, Z.; Long, T.; Wang, Y. KR-12 Coating of Polyetheretherketone (PEEK) Surface: Via Polydopamine Improves Osteointegration and Antibacterial Activity in Vivo. J. Mater. Chem. B 2020, 8, 10190–10204. [Google Scholar] [CrossRef] [PubMed]
- Fabisiak, N.; Fabisiak, A.; Chmielowiec-Korzeniowska, A.; Tymczyna, L.; Kamysz, W.; Kordek, R.; Bauer, M.; Kamysz, E.; Fichna, J. Anti-Inflammatory and Antibacterial Effects of Human Cathelicidin Active Fragment KR-12 in the Mouse Models of Colitis: A Novel Potential Therapy of Inflammatory Bowel Diseases. Pharmacol. Rep. 2021, 73, 163–171. [Google Scholar] [CrossRef]
- Li, H.; Zhang, S.; Nie, B.; Du, Z.; Long, T.; Yue, B. The antimicrobial peptide KR-12 promotes the osteogenic differentiation of human bone marrow stem cells by stimulating BMP/SMAD signaling. RSC Adv. 2018, 8, 15547–15557. [Google Scholar] [CrossRef]
- Luther, A.; Urfer, M.; Zahn, M.; Müller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric Peptidomimetic Antibiotics against Gram-Negative Bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.E.; Hanke, M.L.; Kielian, T. A Mouse Model of Staphylococcus Catheter-Associated Biofilm Infection. Methods Mol. Biol. 2014, 1106, 183–191. [Google Scholar] [CrossRef]
- Xi, L.; Du, J.; Xue, W.; Shao, K.; Jiang, X.; Peng, W.; Li, W.; Huang, S. Cathelicidin LL-37 Promotes Wound Healing in Diabetic Mice by Regulating TFEB-Dependent Autophagy. Peptides 2024, 175, 171183. [Google Scholar] [CrossRef]
- Lee, C.; Kerrigan, C.L.; Picard-Ami, L.A. Cyclophosphamide-Induced Neutropenia: Effect on Postischemic Skin-Flap Survival. Plast. Reconstr. Surg. 1992, 89, 1092–1097. [Google Scholar] [CrossRef]
- Zuluaga, A.F.; Salazar, B.E.; Rodriguez, C.A.; Zapata, A.X.; Agudelo, M.; Vesga, O. Neutropenia Induced in Outbred Mice by a Simplified Low-Dose Cyclophosphamide Regimen: Characterization and Applicability to Diverse Experimental Models of Infectious Diseases. BMC Infect. Dis. 2006, 6, 55. [Google Scholar] [CrossRef]
- Narayana, J.L.; Mishra, B.; Lushnikova, T.; Wu, Q.; Chhonker, Y.S.; Zhang, Y.; Zarena, D.; Salnikov, E.S.; Dang, X.; Wang, F.; et al. Two Distinct Amphipathic Peptide Antibiotics with Systemic Efficacy. Proc. Natl. Acad. Sci. USA 2020, 117, 19446–19454. [Google Scholar] [CrossRef]
- Mishra, B.; Narayana, J.L.; Lushnikova, T.; Wang, X.; Wang, G. Low Cationicity Is Important for Systemic in Vivo Efficacy of Database-Derived Peptides against Drug-Resistant Gram-Positive Pathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 13517–13522. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F.; Arnusch, C.J.; Papahadjopoulos-Sternberg, B.; Wang, G.; Shai, Y. Lipid Clustering by Three Homologous Arginine-Rich Antimicrobial Peptides Is Insensitive to Amino Acid Arrangement and Induced Secondary Structure. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1272–1280. [Google Scholar] [CrossRef]
- Fey, P.D.; Endres, J.L.; Yajjala, V.K.; Widhelm, T.J.; Boissy, R.J.; Bose, J.L.; Bayles, K.W. A Genetic Resource for Rapid and Comprehensive Phenotype Screening of Nonessential Staphylococcus Aureus Genes. mBio 2013, 4, e00537-12. [Google Scholar] [CrossRef]
- Thitirungreangchai, T.; Roytrakul, S.; Aunpad, R. Deciphering the Intracellular Action of the Antimicrobial Peptide A11 via an In-Depth Analysis of Its Effect on the Global Proteome of Acinetobacter Baumannii. ACS Infect. Dis. 2024, 10, 2795–2813. [Google Scholar] [CrossRef] [PubMed]
- Cresti, L.; Cappello, G.; Pini, A. Antimicrobial Peptides towards Clinical Application—A Long History to Be Concluded. Int. J. Mol. Sci. 2024, 25, 4870. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, L.; Fimland, G.; Eijsink, V.; Nissen-Meyer, J. Engineering Increased Stability in the Antimicrobial Peptide Pediocin PA-1. Appl. Environ. Microbiol. 2000, 66, 4798–4802. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Lim, W.H.; Nguyen, P.Q.T.; Tam, J.P. Novel Cyclotides and Uncyclotides with Highly Shortened Precursors from Chassalia Chartacea and Effects of Methionine Oxidation on Bioactivities. J. Biol. Chem. 2012, 287, 17598–17607. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lou, Y.C.; Wang, A.H.J. DMTMM-Mediated Intramolecular Cyclization of Acidic Residues in Peptides/Proteins. ACS Omega 2021, 6, 4708–4718. [Google Scholar] [CrossRef]
- Pang, P.D.; Ahmed, S.M.; Nishiga, M.; Stockbridge, N.L.; Wu, J.C. Tackling the Challenges of New Approach Methods for Predicting Drug Effects from Model Systems. Nat. Rev. Drug Discov. 2024, 23, 565–566. [Google Scholar] [CrossRef]
- Assane, I.M.; Santos-Filho, N.A.; de Sousa, E.L.; de Arruda Brasil, M.C.O.; Cilli, E.M.; Pilarski, F. Cytotoxicity and Antimicrobial Activity of Synthetic Peptides Alone or in Combination with Conventional Antimicrobials against Fish Pathogenic Bacteria. J. Appl. Microbiol. 2021, 131, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Shahriar, S.S.M.; Andrabi, S.M.; Wang, C.; Sharma, N.S.; Xiao, Y.; Wong, S.L.; Wang, G.; Xie, J. It Takes Two to Tangle: Microneedle Patches Co-Delivering Monoclonal Antibodies and Engineered Antimicrobial Peptides Effectively Eradicate Wound Biofilms. Macromol. Biosci. 2024, 24, e2300519. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, M.; Xia, X.; Fu, Y.; Wang, Y.; Xu, W.; Wei, H.; Wei, L. Construction of Exosome-Loaded LL-37 and Its Protection against Zika Virus Infection. Antivir. Res. 2024, 225, 105855. [Google Scholar] [CrossRef] [PubMed]
- John, J.V.; Sharma, N.S.; Tang, G.; Luo, Z.; Su, Y.; Weihs, S.; Shahriar, S.M.S.; Wang, G.; McCarthy, A.; Dyke, J.; et al. Nanofiber Aerogels with Precision Macrochannels and LL-37-Mimic Peptides Synergistically Promote Diabetic Wound Healing. Adv. Funct. Mater. 2023, 33, 2206936. [Google Scholar] [CrossRef]
- Vasconcelos, M.A.; da Silva, B.R.; Andrade, A.L.; de Azevedo Pinheiro, A.; Evaristo, F.F.V.; Arruda, F.V.S.; Lorenzón, E.N.; Cilli, E.M.; Teixeira, E.H. Antimicrobial and Antibiofilm Activity of Synthetic Peptide [W7]KR12-KAEK Against Enterococcus faecalis Strains. Curr. Microbiol. 2023, 80, 325. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Name | Amino Acid Sequence | Comments | Refs. |
|---|---|---|---|---|
| 2008 | KR-12 | KRIVQRIKDFLR-amide | Originally reported KR-12. Short length, narrow-spectrum activity, non-toxic. Need D-amino acids for stability | [32] |
| 2013 | KR-12R | RRIVQRIRDFLR-amide | Enhanced activity of an all-arginine analog | [43] |
| 2013 | KR-12-pa (KR-12-a3) | KRIVKRIKKWLR-amide | Enhanced activity. Need D-amino acids for stability | [44] |
| 2016 | Immobilized KR-12 | CKRIVQRIKDFLR-amide | Immobilized KR-12 to prevent biofilm formation on the titanium surface. Longevity of implants is unknown | [45] |
| 2016 | KR-12/nanofiber | CKRIVQRIKDFLR-amide | Immobilized KR-12 inhibited biofilm formation, enhanced cell attachment, and conferred anti-inflammatory function (for wound healing) | [46] |
| 2017 | KR-12-a5 | KRIVKLILKWLR-amide | Enhanced activity. Need D-amino acids for stability | [47] |
| 2020 | TAT-KR-12 | YGRKKRRQRRRKRIVQRIKDFLR | Added cell penetration ability by increasing peptide length. Need D-amino acids for stability | [48] |
| 2020 | Lipidated KR-12 | C8-KRIVQRIKDFLR-amide | Optimal activity when coupled with the C8 fatty acid | [49] |
| 2021 | Trp cage KR-12 | KRIVQRIKDFLRKYAQWLADGGPSSGRPPPK | KR-12 with stability but longer peptide sequence | [50] |
| 2021 | C10-KR8d (smaller than KR-12) | C10-KRIWQRIK-amide | KR-8 lipopeptide with potency, selectivity, and stability (all D-amino acids) | [51] |
| 2022 | CD4-PP | CPGGKRIVKRIKAFLRGPGGKRIVKRIKAFLR | Head-to-tail cyclic dimeric KR-12. Stable, potent, less selective, increased cost to synthesize | [52] |
| 2023 | CPP-KR12@Si | RKKRRQRRRGSSKRIVQRIKDFLR | Self-trapped CPP-KR12 silica particles with enhanced activity, cell-penetrating ability, increased stability for controlled release. Increased cost of peptide synthesis and cytotoxicity | [53] |
| 2024 | Long-CBP-KR12 | KRIVQRIKDFLRGSGSGGSCQVLNPWYSQTTPGWGQC | KR-12 with carbohydrate-binding property. Costly to synthesize | [54] |
| 2024 | Stapled KR-12 | KRIVQRIKDFLR-amide (i, i+4 Q22-D26) | Stability, selectivity, potency. Can be chemically synthesized | [55] |
| No. | Peptide 1 | MIC | In Vitro Cytotoxicity 2 | Type of Study | In Vivo Model | Treatment Dose | Refs. |
|---|---|---|---|---|---|---|---|
| 1 | Myr-KR-12N | 4–16 μg/mL | RBC: HC50 > 100 μg/mL | 1. Systemic toxicity 2. E. coli induced sepsis model | Mice | 240 μg/mouse | [103] |
| Myr-KR-12C | 8–32 μg/mL | Mice | 10 μg/mouse | [103] | |||
| 2 | KR-12-a2 | 1–8 μM | RBC: HC50 > 800 μM; RAW264.7: LC50 > 100 µM | Topical Ototoxicity | Guinea pigs | 10 μg/mouse | [80,139] |
| 3 | C10-KR8d | 1.5–25 μM | RBC: HC50 > 300 μM; HaCaT: LC50~96 µM | 1. Systemic toxicity | Mice | 40 mg/kg | [51] |
| 2. S. aureus Sepsis model | Mice | 5 mg/kg | |||||
| 3. Catheter-associated biofilm model | Mice | 250 μg/mouse | |||||
| 4 | Hyaluronic acid, Tannic acid, and KR-12 -Cryogel | 3.2 mg of Cryogel | RBC: HC50 > 3.2 mg Cryogel; HaCaT and NIH-3T3 non toxic at 3.2 mg Cryogel | Wound infection model | Mice | 2 mg/mL | [140] |
| 5 | Silk-fibroin-KR-12 | Formulation 20 mg | L929 fibroblasts: LC50~5 μg/mL | Wound infection model | Mice | 2 mg/mL | [141] |
| 6 | LL-37mini | 8–64 μM | RBC: HC50 > 200μM; HaCaT LC50~100 µM | Wound infection model | Mice | 10 mg/kg per wound | [83] |
| 7 | Stapled KR-12 (Q5, D9) | 8–128 μM | RBC: HC50 > 256 μM; NIH-3T3: LC50~168.5 µg/mL | Wound infection model | Mice | 160 μg/mouse | [55] |
| 8 | KR-12-a5 | 1–8 μM | RBC: HC50 > 800 μM; L929 LC50 > 500 ug/mL; RAW264.7: LC50~12.5 µM | LPS-induced bone erosion model | Mice | 2 mg/kg | [142,143] |
| 9 | PEEK-PDA-KR-12 | Formulation with 1 mg/mL | RBMSCs~1 mg formulation | Osteointegration evaluation model | Rats | 1 mg/rat | [144] |
| 10 | KR-12 | 2.5–10 μM | RBC: HC50 > 340 μM; HBMSCs LC50~1000 µM | Induced colitis model | Mice | 5 mg/kg | [51,145,146] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lakshmaiah Narayana, J.; Mechesso, A.F.; Rather, I.I.G.; Zarena, D.; Luo, J.; Xie, J.; Wang, G. Origami of KR-12 Designed Antimicrobial Peptides and Their Potential Applications. Antibiotics 2024, 13, 816. https://doi.org/10.3390/antibiotics13090816
Lakshmaiah Narayana J, Mechesso AF, Rather IIG, Zarena D, Luo J, Xie J, Wang G. Origami of KR-12 Designed Antimicrobial Peptides and Their Potential Applications. Antibiotics. 2024; 13(9):816. https://doi.org/10.3390/antibiotics13090816
Chicago/Turabian StyleLakshmaiah Narayana, Jayaram, Abraham Fikru Mechesso, Imran Ibni Gani Rather, D. Zarena, Jinghui Luo, Jingwei Xie, and Guangshun Wang. 2024. "Origami of KR-12 Designed Antimicrobial Peptides and Their Potential Applications" Antibiotics 13, no. 9: 816. https://doi.org/10.3390/antibiotics13090816