
Innovative Strategies in Drug Repurposing to Tackle Intracellular Bacterial Pathogens

 , , , , , ,
, , , , , ,  and
and 
Abstract
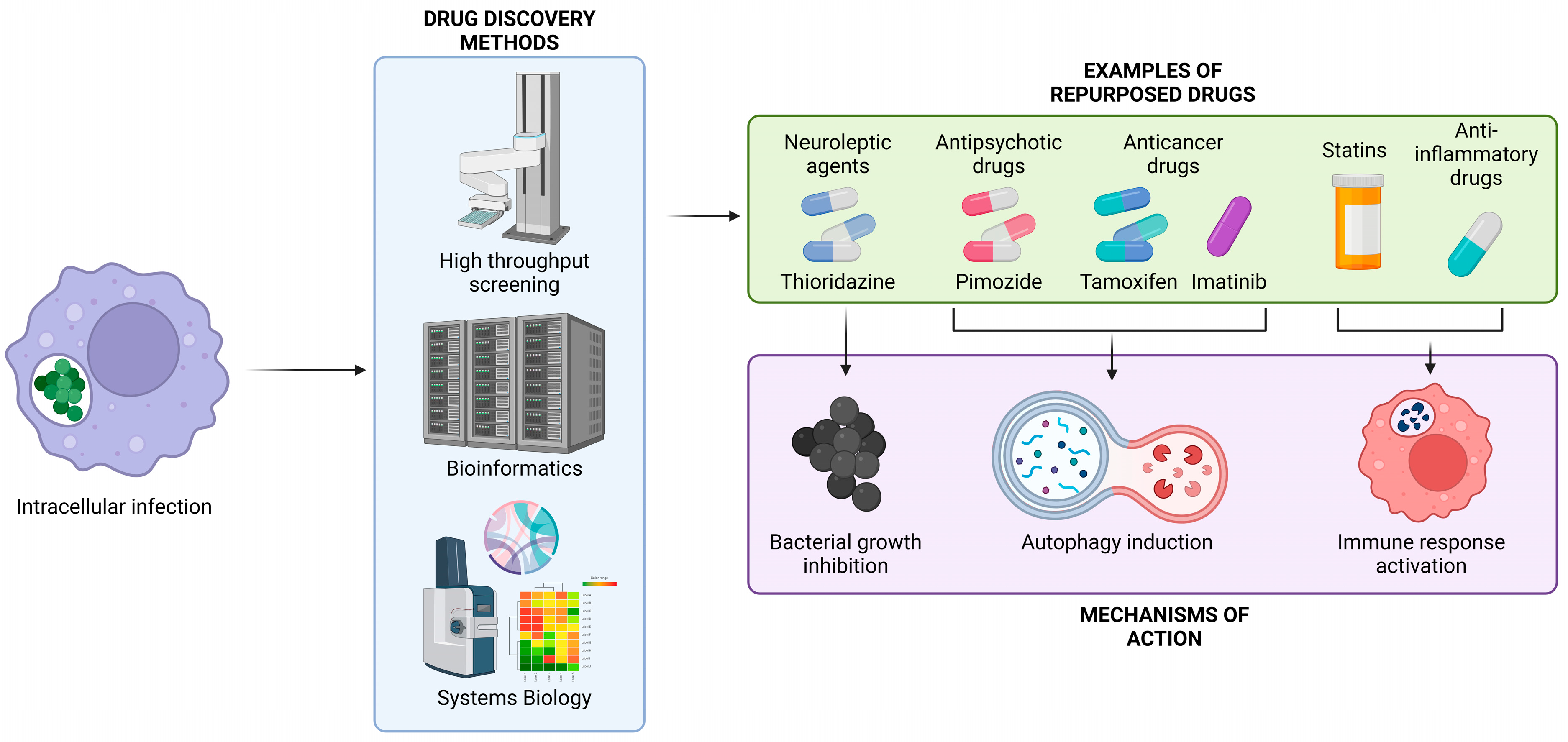
:1. Introduction
2. Staphylococcus aureus

{kind=link}
{kind=link}
| Drug | Type | Mechanism of Action | Reference |
|---|---|---|---|
| Artemisinin | Antimalarial | Generates ROS and DNA damage | [35] |
| Auranofin | Anti-inflammatory | Inhibits thioredoxin reductase | [37] |
| BPH-652 | Cholesterol-lowering | Binds dehydrosqualene synthase (CrtM) | [34] |
| Celecoxib | Anti-inflammatory | Inhibits multidrug efflux pumps | [29] |
| Chlorpromazine | Antipsychotic | Accumulates in lysosomes, triggering antimicrobial activity | [22] |
| Clomiphene | Fertility | Antagonizes wall teichoic acid | [25] |
| Crizotinib | Antitumoral | ATP production | [5,28] |
| Dasatinib | Antitumoral | Tyrosine kinase inhibitor | [15] |
| Diflunisal | Anti-inflammatory | Inhibits the Agr system | [30] |
| Dorsomorphin | AMPK inhibitor | Reduces autophagy and intracellular survival | [43] |
| Ebselen | Anti-inflammatory | Inhibits thioredoxin reductase and thioredoxin | [26] |
| Floxuridine | Antitumoral | Inhibits riboside phosphorylase | [32] |
| Ibrutinib | Antitumoral | Controls the MEK/ERK/c-JUN signaling pathway | [15] |
| Phenothiazine | Antipsychotic | Antagonism of dopamine D2 receptors | [11,13] |
| Raloxifene | Estrogen receptor agonist | Induces autophagy and inhibits neutrophil cell death | [41] |
| Selamectin | Anthelmintic | Shows high activity against S. aureus and M. tuberculosis | [28,41] |
| Streptozotocin | Antitumoral | DNA synthesis inhibitor | [28,32] |
| Thapsigargin | Ca2+ endoplasmic pump inhibitor | Increases host cell viability and reduces intracellular survival | [16,32] |
| Thioridazine | Antipsychotic | Accumulates in macrophages, triggering antimicrobial activity | [16,22,23] |
3. Mycobacterium tuberculosis
| Drug | Type | Mechanism of Action | Reference |
|---|---|---|---|
| Artemisinin | Antimalarial | Generates ROS and DNA damage | [74] |
| Artesunate | Antimalarial | Unknown | [74] |
| Atorvastatin | Statin | Enhances macrophage bactericidal effects | [50] |
| Auranofin | Anti-inflammatory | Inhibits thioredoxin reductase | [37] |
| Bazedoxifene | Estrogen receptor modulator | Reduces intracellular growth | [59] |
| Bevacizumab | Antitumoral | Neutralizes VEGF | [65] |
| Biapenem | Antimicrobial | Targets cell wall synthesis | [72] |
| Chlorpromazine | Antipsychotic | NADH dehydrogenase type II inhibitor | [76] |
| Faropenem | Antimicrobial | Targets cell wall synthesis | [72] |
| Fluspirilene | Antipsychotic | Elicits autophagy | [56] |
| Fluvastatin | Statin | Targets intracellular activity | [68] |
| Gatifloxacin | Antimicrobial | DNA gyrase | [76] |
| Ibuprofen | Anti-inflammatory | Reduces inflammatory response | [53] |
| Ibrutinib | Antitumoral | Inhibits the BTK/Akt/mTOR pathway, triggering autophagy | [39] |
| Imatinib | Antitumoral | Inhibits tyrosine kinases | [57] |
| Imidazopyridine amides (Q203) | Antimicrobial | Cytochrome bc1 complex | [61] |
| Ivermectin | Anthelmintic | Inhibits intracellular growth | [69] |
| Metformin | Antidiabetic | Activates energy-sensing kinase and reduces inflammation | [63] |
| Moxidectin | Anthelmintic | Unknown mechanism of action | [55] |
| Nitroimidazofuran (PA-824) | Antimicrobial | Bacterial nitroreduction | [49] |
| Nitazoxanide | Anthelmintic | Modulates host immune responses | [70] |
| Phenothiazine | Antipsychotic | NADH inhibitor | [57,62] |
| Pimozide | Antipsychotic | Affects ROS generation and inhibits STAT5 | [56] |
| Saquinavir | Antiviral | Restores cathepsin protease activity | [48] |
| Selamectin | Anthelmintic | Unknown mechanism of action | [55] |
| Simvastatin | Statin | Reduces cholesterol uptake, showing synergy with antimicrobials | [51] |
| Sulfadiazine | Antimicrobial | Inhibits folic acid synthesis | [66] |
| Tamoxifen | Antitumoral | Induces autophagy | [55] |
| Tebipenem | Antimicrobial | Targets cell wall synthesis | [72] |
| Terlipressin | Vasoactive | Targets EmbC for cell wall synthesis | [67] |
| Verapamil | Cardiovascular | Inhibits bacterial efflux pump | [39] |
| Vitamin D | Vitamin | Activates TLR and induces autophagy | [54] |
4. Listeria monocytogenes
5. Salmonella enterica Serovar Typhimurium
| Drug | Type | Mechanism of Action | Reference |
|---|---|---|---|
| Amoxapine | Antidepressant | Increase the levels of norepinephrine and serotonin | [74] |
| Amenamevir | Antiviral | Inhibits helicase-primase complex | [75] |
| Azacitidine | Antitumoral | Disrupts cytoplasmic membrane potential | [84] |
| Bromperidol | Antipsychotic | Dopamine D2 receptor antagonist | [70] |
| Carmofur | Antitumoral | Inhibitor dihydropyrimidine dehydrogenase | [84] |
| Ciclopirox | Antifungal | Inhibits iron-dependent enzymes | [70] |
| Diclofenac sodium | Anti-inflammatory | Inhibits prostaglandin G/H synthase | [74] |
| Doxapram | Respiratory stimulant | Stimulates respiratory chemoreceptors | [74] |
| Doxifluridine | Antitumoral | Inhibits thymidylate synthase | [70] |
| Duvelisib | Antitumoral | Inhibits δ and γ isoforms PI3K | [75] |
| Ethopropazine | Anticholinergic | Blocks muscarinic acetylcholine receptors | [70] |
| Fluorouracil | Antitumoral | Disrupts cytoplasmic membrane potential | [84] |
| Lifitegrast | Anti-inflammatory | Integrin agonist | [75] |
| Loperamide | Antidiarrheal | Promotes autophagy | [89] |
| Lovastatin | Anti-cholesterol | Inhibits HMG-CoA reductase | [72] |
| Metergoline | Antipsychotic | Serotonin receptor antagonist | [70] |
| Nilotinib | Antitumoral | Inhibits tyrosine kinase | [75] |
| Trifluoperazine | Antipsychotic | Accumulates in macrophages, targets autophagy pathway | [88] |
6. Other Intracellular Pathogens
| Drug | Type | Mechanism of Action | Pathogen | Reference |
|---|---|---|---|---|
| Capsaicin | Herbal compound | Induces autophagy | S. flexneri | [91] |
| Diclofenac sodium | Anti-inflammatory | Inhibits prostaglandin G/H synthase | Shigella sp. | [87] |
| Doxapram | Respiratory | Blocks intracellular colonization | Y. pestis | [88] |
| Simvastatin | Statin | HMG-CoA inhibitor | C. pneumoniae | [94] |
| Trifluoperazine | Antipsychotic | Accumulates in macrophages, targets autophagy pathway | C. pneumoniae, C. difficile, Y. pestis | [88] |
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J. Immunol. Res. 2019, 2019, 1356540. [Google Scholar] [CrossRef] [PubMed]
- Leseigneur, C.; Le-Bury, P.; Pizarro-Cerda, J.; Dussurget, O. Emerging Evasion Mechanisms of Macrophage Defenses by Pathogenic Bacteria. Front. Cell Infect. Microbiol. 2020, 10, 577559. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.; Grigsby, S.J.; Philips, J.A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2022, 20, 750–766. [Google Scholar] [CrossRef] [PubMed]
- Shapira, T.; Christofferson, M.; Av-Gay, Y. The antimicrobial activity of innate host-directed therapies: A systematic review. Int. J. Antimicrob. Agents 2024, 63, 107138. [Google Scholar] [CrossRef] [PubMed]
- Konreddy, A.K.; Rani, G.U.; Lee, K.; Choi, Y. Recent Drug-Repurposing-Driven Advances in the Discovery of Novel Antibiotics. Curr. Med. Chem. 2019, 26, 5363–5388. [Google Scholar] [CrossRef]
- Quezada, H.; Martinez-Vazquez, M.; Lopez-Jacome, E.; Gonzalez-Pedrajo, B.; Andrade, A.; Fernandez-Presas, A.M.; Tovar-Garcia, A.; Garcia-Contreras, R. Repurposed anti-cancer drugs: The future for anti-infective therapy? Expert. Rev. Anti Infect. Ther. 2020, 18, 609–612. [Google Scholar] [CrossRef]
- Ahmad, Z.; Rauf, A.; Naz, S.; Hemeg, H.A. Introduction to Drug Repurposing: Exploring New Applications for Existing Drugs; InTech Open: London, UK, 2024. [Google Scholar]
- Galindez, G.; Matschinske, J.; Rose, T.D.; Sadegh, S.; Salgado-Albarran, M.; Spath, J.; Baumbach, J.; Pauling, J.K. Lessons from the COVID-19 pandemic for advancing computational drug repurposing strategies. Nat. Comput. Sci. 2021, 1, 33–41. [Google Scholar] [CrossRef]
- Kulkarni, V.S.; Alagarsamy, V.; Solomon, V.R.; Jose, P.A.; Murugesan, S. Drug Repurposing: An Effective Tool in Modern Drug Discovery. Russ. J. Bioorg. Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Juarez-Lopez, D.; Schcolnik-Cabrera, A. Drug Repurposing: Considerations to Surpass While Re-directing Old Compounds for New Treatments. Arch. Med. Res. 2021, 52, 243–251. [Google Scholar] [CrossRef]
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, e183–e198. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J. Drug repurposing to overcome microbial resistance. Drug Discov. Today 2022, 27, 2028–2041. [Google Scholar] [CrossRef] [PubMed]
- Mourenza, A.; Gil, J.A.; Mateos, L.M.; Letek, M. Novel Treatments against Mycobacterium tuberculosis Based on Drug Repurposing. Antibiotics 2020, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Santano, N.; Stolting, H.; Cooper, F.; Bileckaja, N.; Majstorovic, A.; Ihle, N.; Mateos, L.M.; Calle, Y.; Behrends, V.; Letek, M. Host-directed kinase inhibitors act as novel therapies against intracellular Staphylococcus aureus. Sci. Rep. 2019, 9, 4876. [Google Scholar] [CrossRef]
- Bravo-Santano, N.; Capilla-Lasheras, P.; Mateos, L.M.; Calle, Y.; Behrends, V.; Letek, M. Identification of novel targets for host-directed therapeutics against intracellular Staphylococcus aureus. Sci. Rep. 2019, 9, 15435. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Hajam, I.A.; Liu, G.Y. Linking S. aureus Immune Evasion Mechanisms to Staphylococcal Vaccine Failures. Antibiotics 2024, 13, 410. [Google Scholar] [CrossRef]
- Yu, H.; Xu, Y.; Imani, S.; Zhao, Z.; Ullah, S.; Wang, Q. Navigating ESKAPE Pathogens: Considerations and Caveats for Animal Infection Models Development. ACS Infect. Dis. 2024, 10, 2336–2355. [Google Scholar] [CrossRef]
- Das, S.; Dasgupta, A.; Chopra, S. Drug repurposing: A new front in the war against Staphylococcus aureus. Future Microbiol. 2016, 11, 1091–1099. [Google Scholar] [CrossRef]
- Martins, M.; Bleiss, W.; Marko, A.; Ordway, D.; Viveiros, M.; Leandro, C.; Pacheco, T.; Molnar, J.; Kristiansen, J.E.; Amaral, l. Clinical Concentrations of Thioridazine Enhance the Killing of Intracellular Methicillin-resistant Staphylococcus aureus: An In Vivo, Ex Vivo and Electron Microscopy Study. In Vivo 2004, 18, 787–794. [Google Scholar] [PubMed]
- Amaral, L.; Kristiansen, J.E.; Abebe, L.S.; Millett, W. Inhibition of the respiration of multi-drug resistant clinical isolates of Mycobacterium tuberculosis by thioridazine: Potential use for initial therapy of freshly diagnosed tuberculosis. J. Antimicrob. Chemother. 1996, 38, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Ordwaya, D.; Viveiros, M.; Leandro, C.; Arroz, M.J.; Amaral, L. Intracellular activity of clinical concentrations of phenothiazines including thioridiazine against phagocytosed Staphylococcus aureus. Int. J. Antimicrob. Agents 2002, 20, 34–43. [Google Scholar] [CrossRef]
- Farhaa, M.A.; Czarnya, T.L.; Myersa, C.L.; Worrallc, L.J.; Frencha, S.; Conradyc, D.G.; Wangd, Y.; Oldfieldd, E.; Strynadkac, N.C.J.; Brown, E.D. Antagonism screen for inhibitors of bacterial cell wall biogenesis uncovers an inhibitor of undecaprenyl diphosphate synthase. Proc. Natl. Acad. Sci. USA 2015, 112, 11048–11053. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Li, C.; Chen, Y.; Deng, Y.; Yang, T.; Luo, Y. Repurposed drug candidates for antituberculosis therapy. Eur. J. Med. Chem. 2020, 192, 112175. [Google Scholar] [CrossRef]
- Thangamani, S.; Younis, W.; Seleem, M.N. Repurposing Clinical Molecule Ebselen to Combat Drug Resistant Pathogens. PLoS ONE 2015, 10, e0133877. [Google Scholar] [CrossRef]
- Folliero, V.; Dell’Annunziata, F.; Santella, B.; Roscetto, E.; Zannella, C.; Capuano, N.; Perrella, A.; De Filippis, A.; Boccia, G.; Catania, M.R.; et al. Repurposing Selamectin as an Antimicrobial Drug against Hospital-Acquired Staphylococcus aureus Infections. Microorganisms 2023, 11, 2242. [Google Scholar] [CrossRef]
- Thangamani, S.; Younis, W.; Seleem, M.N. Repurposing celecoxib as a topical antimicrobial agent. Front. Microbiol. 2015, 6, 750. [Google Scholar] [CrossRef]
- Hendrix, A.S.; Spoonmore, T.J.; Wilde, A.D.; Putnam, N.E.; Hammer, N.D.; Snyder, D.J.; Guelcher, S.A.; Skaar, E.P.; Cassat, J.E. Repurposing the Nonsteroidal Anti-inflammatory Drug Diflunisal as an Osteoprotective, Antivirulence Therapy for Staphylococcus aureus Osteomyelitis. Antimicrob. Agents Chemother. 2016, 60, 5322–5330. [Google Scholar] [CrossRef]
- Chifiriuc, M.C.; Filip, R.; Constantin, M.; Pircalabioru, G.G.; Bleotu, C.; Burlibasa, L.; Ionica, E.; Corcionivoschi, N.; Mihaescu, G. Common themes in antimicrobial and anticancer drug resistance. Front. Microbiol. 2022, 13, 960693. [Google Scholar] [CrossRef]
- Yeo, W.S.; Arya, R.; Kim, K.K.; Jeong, H.; Cho, K.H.; Bae, T. The FDA-approved anti-cancer drugs, streptozotocin and floxuridine, reduce the virulence of Staphylococcus aureus. Sci. Rep. 2018, 8, 2521. [Google Scholar] [CrossRef] [PubMed]
- Linzner, N.; Loi, V.V.; Antelmann, H. The Catalase KatA Contributes to Microaerophilic H2O2 Priming to Acquire an Improved Oxidative Stress Resistance in Staphylococcus aureus. Antioxidants 2022, 11, 1793. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.I.; Liu, G.Y.; Song, Y.; Yin, F.; Hensler, M.E.; Jeng, W.Y.; Nizet, V.; Wang, A.H.; Oldfield, E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. [Google Scholar] [CrossRef]
- Chung, I.Y.; Jang, H.J.; Yoo, Y.J.; Hur, J.; Oh, H.Y.; Kim, S.H.; Cho, Y.H. Artemisinin displays bactericidal activity via copper-mediated DNA damage. Virulence 2022, 13, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Vlamis-Gardikas, A.; Kandasamy, K.; Zhao, R.; Gustafsson, T.N.; Engstrand, L.; Hoffner, S.; Engman, L.; Holmgren, A. Inhibition of bacterial thioredoxin reductase: An antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 2013, 27, 1394–1403. [Google Scholar] [CrossRef]
- Harbut, M.B.; Vilcheze, C.; Luo, X.; Hensler, M.E.; Guo, H.; Yang, B.; Chatterjee, A.K.; Nizet, V.; Jacobs, W.R., Jr.; Schultz, P.G.; et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 4453–4458. [Google Scholar] [CrossRef]
- Thangamani, S.; Mohammad, H.; Abushahba, M.F.; Sobreira, T.J.; Hedrick, V.E.; Paul, L.N.; Seleem, M.N. Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci. Rep. 2016, 6, 22571. [Google Scholar] [CrossRef]
- Hu, Y.; Wen, Z.; Liu, S.; Cai, Y.; Guo, J.; Xu, Y.; Lin, D.; Zhu, J.; Li, D.; Chen, X. Ibrutinib suppresses intracellular Mycobacterium tuberculosis growth by inducing macrophage autophagy. J. Infect. 2020, 80, e19–e26. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhong, T.; Wu, H.; Li, N.; Fang, Z.; Cao, L.; Yin, X.; He, Q.; Ge, R.; Sun, X. Crizotinib Shows Antibacterial Activity against Gram-Positive Bacteria by Reducing ATP Production and Targeting the CTP Synthase PyrG. Microbiol. Spectr. 2022, 10, e00884-22. [Google Scholar] [CrossRef]
- Chang, J.; Kim, J.; Lee, W. Raloxifene prevents intracellular invasion of pathogenic bacteria through modulation of cell metabolic pathways. J. Antimicrob. Chemother. 2022, 77, 1617–1624. [Google Scholar] [CrossRef]
- Flores, R.; Dohrmann, S.; Schaal, C.; Hakkim, A.; Nizet, V.; Corriden, R. The Selective Estrogen Receptor Modulator Raloxifene Inhibits Neutrophil Extracellular Trap Formation. Front. Immunol. 2016, 7, 566. [Google Scholar] [CrossRef]
- Bravo-Santano, N.; Ellis, J.K.; Mateos, L.M.; Calle, Y.; Keun, H.C.; Behrends, V.; Letek, M. Intracellular Staphylococcus aureus Modulates Host Central Carbon Metabolism To Activate Autophagy. mSphere 2018, 3, e00374-18. [Google Scholar] [CrossRef] [PubMed]
- Villar-Hernandez, R.; Ghodousi, A.; Konstantynovska, O.; Duarte, R.; Lange, C.; Raviglione, M. Tuberculosis: Current challenges and beyond. Breathe 2023, 19, 220166. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Maeurer, M.; Host-Directed Therapies, N.; Chakaya, J.; Hoelscher, M.; Ntoumi, F.; Rustomjee, R.; Vilaplana, C.; Yeboah-Manu, D.; Rasolof, V.; et al. Towards host-directed therapies for tuberculosis. Nat. Rev. Drug Discov. 2015, 14, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Torfs, E.; Piller, T.; Cos, P.; Cappoen, D. Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy. Int. J. Mol. Sci. 2019, 20, 2868. [Google Scholar] [CrossRef]
- Kumar, A.; Alam, A.; Grover, S.; Pandey, S.; Tripathi, D.; Kumari, M.; Rani, M.; Singh, A.; Akhter, Y.; Ehtesham, N.Z.; et al. Peptidyl-prolyl isomerase-B is involved in Mycobacterium tuberculosis biofilm formation and a generic target for drug repurposing-based intervention. NPJ Biofilms Microbiomes 2019, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, J.M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium tuberculosis Infection. Front. Immunol. 2021, 12, 647728. [Google Scholar] [CrossRef] [PubMed]
- de Chastellier, C.; Thilo, L. Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome-derived autophagic vacuoles. Cell Microbiol. 2006, 8, 242–256. [Google Scholar] [CrossRef]
- Lobato, L.S.; Rosa, P.S.; da Silva Ferreira, J.; da Silva Neumann, A.; Gomes da Silva, M.; Caitano do Nascimento, D.; Teixeira Soares, C.; Barbosa Pedrini, S.C.; Leal de Oliveira, D.S.; Peres Monteiro, C.; et al. Statins Increase Rifampin Mycobactericidal Effect. Antimicrob. Agents Chemother. 2014, 58, 5766–5774. [Google Scholar] [CrossRef]
- Skerry, C.; Pinn, M.L.; Bruiners, N.; Pine, R.; Gennaro, M.L.; Karakousis, P.C. Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J. Antimicrob. Chemother. 2014, 69, 2453–2457. [Google Scholar] [CrossRef]
- Gupta, S.; Tyagi, S.; Almeida, D.V.; Maiga, M.C.; Ammerman, N.C.; Bishai, W.R. Acceleration of tuberculosis treatment by adjunctive therapy with verapamil as an efflux inhibitor. Am. J. Respir. Crit. Care Med. 2013, 188, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Vilaplana, C.; Marzo, E.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.J. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J. Infect. Dis. 2013, 208, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-Like Receptor Triggering of a Vitamin D–Mediated Human Antimicrobial Response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Boland, R.; Heemskerk, M.T.; Forn-Cuní, G.; Korbee, C.J.; Walburg, K.V.; Esselink, J.J.; Carvalho dos Santos, C.; de Waal, A.M.; van der Hoeven, D.C.M.; van der Sar, E.; et al. Repurposing Tamoxifen as Potential Host-Directed Therapeutic for Tuberculosis. mBio 2022, 14, e0302422. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, M.T.; Korbee, C.J.; Esselink, J.J.; Dos Santos, C.C.; van Veen, S.; Gordijn, I.F.; Vrieling, F.; Walburg, K.V.; Engele, C.G.; Dijkman, K.; et al. Repurposing diphenylbutylpiperidine-class antipsychotic drugs for host-directed therapy of Mycobacterium tuberculosis and Salmonella enterica infections. Sci. Rep. 2021, 11, 19634. [Google Scholar] [CrossRef] [PubMed]
- Napier, R.J.; Rafi, W.; Cheruvu, M.; Powell, K.R.; Zaunbrecher, M.A.; Bornmann, W.; Salgame, P.; Shinnick, T.M.; Kalman, D. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe 2011, 10, 475–485. [Google Scholar] [CrossRef]
- Kuijl, C.; Savage, N.D.; Marsman, M.; Tuin, A.W.; Janssen, L.; Egan, D.A.; Ketema, M.; van den Nieuwendijk, R.; van den Eeden, S.J.; Geluk, A.; et al. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 2007, 450, 725–730. [Google Scholar] [CrossRef]
- Ouyang, Q.; Zhang, K.; Lin, D.; Feng, C.G.; Cai, Y.; Chen, X. Bazedoxifene Suppresses Intracellular Mycobacterium tuberculosis Growth by Enhancing Autophagy. mSphere 2020, 5, e00124-20. [Google Scholar] [CrossRef]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Yang, C.S.; Jin, H.S.; Kim, K.K.; Lee, Z.W.; Lee, S.H.; Kim, J.M.; Jo, E.K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I. Metformin: Candidate host-directed therapy for tuberculosis in diabetes and non-diabetes patients. Tuberculosis 2016, 101S, S69–S72. [Google Scholar] [CrossRef] [PubMed]
- Magee, M.J.; Salindri, A.D.; Kornfeld, H.; Singhal, A. Reduced prevalence of latent tuberculosis infection in diabetes patients using metformin and statins. Eur. Respir. J. 2019, 53, 1801695. [Google Scholar] [CrossRef]
- Datta, M.; Via, L.E.; Kamoun, W.S.; Liu, C.; Chen, W.; Seano, G.; Weiner, D.M.; Schimel, D.; England, K.; Martin, J.D.; et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc. Natl. Acad. Sci. USA 2015, 112, 1827–1832. [Google Scholar] [CrossRef]
- Ameen, S.M.; Drancourt, M. In vitro susceptibility of Mycobacterium tuberculosis to trimethoprim and sulfonamides in France. Antimicrob. Agents Chemother. 2013, 57, 6370–6371. [Google Scholar] [CrossRef]
- Singh, A.; Somvanshi, P.; Grover, A. Drug repurposing against arabinosyl transferase (EmbC) of Mycobacterium tuberculosis: Essential dynamics and free energy minima based binding mechanics analysis. Gene 2019, 693, 114–126. [Google Scholar] [CrossRef]
- Battah, B.; Chemi, G.; Butini, S.; Campiani, G.; Brogi, S.; Delogu, G.; Gemma, S. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles. Molecules 2019, 24, 4373. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.E.; Vilcheze, C.; Ng, C.; Jacobs, W.R., Jr.; Ramon-Garcia, S.; Thompson, C.J. Anthelmintic avermectins kill Mycobacterium tuberculosis, including multidrug-resistant clinical strains. Antimicrob. Agents Chemother. 2013, 57, 1040–1046. [Google Scholar] [CrossRef]
- Ranjbar, S.; Haridas, V.; Nambu, A.; Jasenosky, L.D.; Sadhukhan, S.; Ebert, T.S.; Hornung, V.; Cassell, G.H.; Falvo, J.V.; Goldfeld, A.E. Cytoplasmic RNA Sensor Pathways and Nitazoxanide Broadly Inhibit Intracellular Mycobacterium tuberculosis Growth. iScience 2019, 22, 299–313. [Google Scholar] [CrossRef]
- Salie, S.; Labuschagne, A.; Walters, A.; Geyer, S.; Jardine, A.; Jacobs, M.; Hsu, N.J. In vitro and in vivo toxicity evaluation of non-neuroleptic phenothiazines, antitubercular drug candidates. Regul. Toxicol. Pharmacol. 2019, 109, 104508. [Google Scholar] [CrossRef]
- Kumar, P.; Kaushik, A.; Lloyd, E.P.; Li, S.G.; Mattoo, R.; Ammerman, N.C.; Bell, D.T.; Perryman, A.L.; Zandi, T.A.; Ekins, S.; et al. Non-classical transpeptidases yield insight into new antibacterials. Nat. Chem. Biol. 2017, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Makkar, N.; Pandey, P.; Parrish, N.; Singh, U.; Lamichhane, G. Carbapenems and Rifampin Exhibit Synergy against Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob. Agents Chemother. 2015, 59, 6561–6567. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.H. Novel Pharmacological Activity of Artesunate and Artemisinin: Their Potential as Anti-Tubercular Agents. J. Clin. Med. 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ahmed, F.; Sharma, T.; Grover, A.; Agarwal, M.; Grover, S. Potential Repurposed Drug Candidates for Tuberculosis Treatment: Progress and Update of Drugs Identified in Over a Decade. ACS Omega 2023, 8, 17362–17380. [Google Scholar] [CrossRef]
- Schuchat, A.; Swaminathan, B.; Broome, C.V. Epidemiology of Human Listeriosis. Clin. Microbiol. Rev. 1991, 4, 169–183. [Google Scholar] [CrossRef]
- Cong, Z.; Xiong, Y.; Lyu, L.; Fu, B.; Guo, D.; Sha, Z.; Yang, B.; Wu, H. The relationship between Listeria infections and host immune responses: Listeriolysin O as a potential target. Biomed. Pharmacother. 2024, 171, 116129. [Google Scholar] [CrossRef]
- Beauregard, K.E.; Lee, K.-D.; Collier, R.J.; Swanson, J.A. pH-dependent Perforation of Macrophage Phagosomes by Listeriolysin O from Listeria monocytogenes. J. Exp. Med. 1997, 186, 1159–1163. [Google Scholar] [CrossRef]
- Gonzales-Barron, U.; Cadavez, V.; Guillier, L.; Sanaa, M. A Critical Review of Risk Assessment Models for Listeria monocytogenes in Dairy Products. Foods 2023, 12, 4436. [Google Scholar] [CrossRef]
- Lieberman, L.A.; Higgins, D.E. Inhibition of Listeria monocytogenes infection by neurological drugs. Int. J. Antimicrob. Agents 2010, 35, 292–296. [Google Scholar] [CrossRef]
- Lieberman, L.A.; Higgins, D.E. A small-molecule screen identifies the antipsychotic drug pimozide as an inhibitor of Listeria monocytogenes infection. Antimicrob. Agents Chemother. 2009, 53, 756–764. [Google Scholar] [CrossRef]
- Geronikaki, A.; Kartsev, V.; Petrou, A.; Akrivou, M.G.; Vizirianakis, I.S.; Chatzopoulou, F.M.; Lichitsky, B.; Sirakanyan, S.; Kostic, M.; Smiljkovic, M.; et al. Antibacterial activity of griseofulvin analogues as an example of drug repurposing. Int. J. Antimicrob. Agents 2020, 55, 105884. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Tsai, C.N.; Johnson, J.W.; French, S.; Elhenawy, W.; Porwollik, S.; Andrews-Polymenis, H.; McClelland, M.; Magolan, J.; Coombes, B.K.; et al. A macrophage-based screen identifies antibacterial compounds selective for intracellular Salmonella Typhimurium. Nat. Commun. 2019, 10, 197. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ren, Q.; Ni, T.; Zhao, Y.; Sang, Z.; Luo, R.; Li, Z.; Li, S. Strategies adopted by Salmonella to survive in host: A review. Arch. Microbiol. 2023, 205, 362. [Google Scholar] [CrossRef] [PubMed]
- Catron, D.M.; Lange, Y.; Borensztajn, J.; Sylvester, M.D.; Jones, B.D.; Haldar, K. Salmonella enterica serovar Typhimurium requires nonsterol precursors of the cholesterol biosynthetic pathway for intracellular proliferation. Infect. Immun. 2004, 72, 1036–1042. [Google Scholar] [CrossRef]
- Dutta, N.K.; Annadurai, S.; Mazumdar, K.; Dastidar, S.G.; Kristiansen, J.E.; Molnar, J.; Martins, M.; Amaral, L. Potential management of resistant microbial infections with a novel non-antibiotic: The anti-inflammatory drug diclofenac sodium. Int. J. Antimicrob. Agents 2007, 30, 242–249. [Google Scholar] [CrossRef]
- Andersson, J.A.; Fitts, E.C.; Kirtley, M.L.; Ponnusamy, D.; Peniche, A.G.; Dann, S.M.; Motin, V.L.; Chauhan, S.; Rosenzweig, J.A.; Sha, J.; et al. New Role for FDA-Approved Drugs in Combating Antibiotic-Resistant Bacteria. Antimicrob. Agents Chemother. 2016, 60, 3717–3729. [Google Scholar] [CrossRef]
- Liu, H.; Li, S.; Denga, L.; Shi, Z.; Jiang, C.; Shua, J.; Liu, Y.; Denga, X.; Wanga, J.; Guoa, Z.; et al. Repurposing Loperamide as an Anti-Infection Drug for the Treatment of Intracellular Bacterial Pathogens. Engineering 2024. [Google Scholar] [CrossRef]
- Joshi, T.; Sharma, P.; Joshi, T.; Mathpal, S.; Pande, V.; Chandra, S. Repurposing of FDA approved drugs against Salmonella enterica serovar Typhi by targeting dihydrofolate reductase: An in silico study. J. Biomol. Struct. Dyn. 2022, 40, 3731–3744. [Google Scholar] [CrossRef]
- Basak, P.; Maitra, P.; Khan, U.; Saha, K.; Bhattacharya, S.S.; Dutta, M.; Bhattacharya, S. Capsaicin Inhibits Shigella flexneri Intracellular Growth by Inducing Autophagy. Front. Pharmacol. 2022, 13, 903438. [Google Scholar] [CrossRef]
- Kronenberg, A.; Butikofer, L.; Odutayo, A.; Muhlemann, K.; da Costa, B.R.; Battaglia, M.; Meli, D.N.; Frey, P.; Limacher, A.; Reichenbach, S.; et al. Symptomatic treatment of uncomplicated lower urinary tract infections in the ambulatory setting: Randomised, double blind trial. BMJ 2017, 359, j4784. [Google Scholar] [CrossRef] [PubMed]
- Czyz, D.M.; Potluri, L.P.; Jain-Gupta, N.; Riley, S.P.; Martinez, J.J.; Steck, T.L.; Crosson, S.; Shuman, H.A.; Gabay, J.E. Host-directed antimicrobial drugs with broad-spectrum efficacy against intracellular bacterial pathogens. mBio 2014, 5, e01534-14. [Google Scholar] [CrossRef] [PubMed]
- Erkkila, L.; Jauhiainen, M.; Laitinen, K.; Haasio, K.; Tiirola, T.; Saikku, P.; Leinonen, M. Effect of simvastatin, an established lipid-lowering drug, on pulmonary Chlamydia pneumoniae infection in mice. Antimicrob. Agents Chemother. 2005, 49, 3959–3962. [Google Scholar] [CrossRef] [PubMed]

| Drug | Type | Mechanism of Action | Reference |
|---|---|---|---|
| Bepridil | Antitumoral | Inhibits calcium channel inhibitor | [67] |
| Griseofulvin | Antifungal | Disruptor of microtubules | [69] |
| Pimozide | Antipsychotic | Dopamine D2 receptor antagonist | [68] |
| Thioridazine | Antipsychotic | Dopamine D2 receptor antagonist | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorente-Torres, B.; Llano-Verdeja, J.; Castañera, P.; Ferrero, H.Á.; Fernández-Martínez, S.; Javadimarand, F.; Mateos, L.M.; Letek, M.; Mourenza, Á. Innovative Strategies in Drug Repurposing to Tackle Intracellular Bacterial Pathogens. Antibiotics 2024, 13, 834. https://doi.org/10.3390/antibiotics13090834
Lorente-Torres B, Llano-Verdeja J, Castañera P, Ferrero HÁ, Fernández-Martínez S, Javadimarand F, Mateos LM, Letek M, Mourenza Á. Innovative Strategies in Drug Repurposing to Tackle Intracellular Bacterial Pathogens. Antibiotics. 2024; 13(9):834. https://doi.org/10.3390/antibiotics13090834
Chicago/Turabian StyleLorente-Torres, Blanca, Jesús Llano-Verdeja, Pablo Castañera, Helena Á. Ferrero, Sergio Fernández-Martínez, Farzaneh Javadimarand, Luis M. Mateos, Michal Letek, and Álvaro Mourenza. 2024. "Innovative Strategies in Drug Repurposing to Tackle Intracellular Bacterial Pathogens" Antibiotics 13, no. 9: 834. https://doi.org/10.3390/antibiotics13090834