Abstract
Staphylococcus aureus is a bacterial species that is commonly found colonising healthy individuals but that presents a paradoxical nature: simultaneously, it can migrate within the body and cause a range of diseases. Many of these become chronic by resisting immune responses, antimicrobial treatment, and medical intervention. In part, this ability to persist can be attributed to the adoption of multiple cell types within a single cellular population. These dynamics in the S. aureus cell population could be the result of its interplay with host cells or other co-colonising bacteria—often coagulase-negative Staphylococcal (CoNS) species. Further understanding of the unique traits of S. aureus alternative cell types, the drivers for their selection or formation during disease, as well as their presence even during non-pathological colonisation could advance the development of diagnostic tools and drugs tailored to target specific cells that are eventually responsible for chronic infections.
1. Introduction
Staphylococcus aureus is one of the most notorious bacterial species that are globally responsible for significant infection-related morbidity and mortality, with a fatality burden that consistently exceeds over one million deaths annually [1]. This reality is an unfortunate consequence of S. aureus’s ability to cause acute and chronic infections presented across a broad spectrum of diseases, from common skin and soft tissue infections to the more severe bacteraemia, pneumonia, osteomyelitis (OM), complications with cystic fibrosis (CF), and endocarditis, which can all lead to sepsis or death [2]. However, S. aureus’s success as a microorganism is not just a result of its ability to cause disease; instead, it can be largely attributed to S. aureus’s remarkable ability to adapt and colonise a range of anatomical niches within a human host [3]. S. aureus has been found to intermittently colonise approximately 30% of the healthy human population, predominantly in the mucous membrane, anterior nares, and the skin, and possibly also in the gut [4]. Although these colonising strains of S. aureus are asymptomatic, their opportunistic nature causes the bacteria to concurrently possess the ability to invade other sites in the body and cause disease to its human host, often when the immune system is compromised [5]. Colonising S. aureus contributes to the infection rates caused by community-acquired and nosocomial infections, as well as causes endogenous infections where the bacteria can transit through its host [6]. S. aureus’s prevalence and ability to become a pathogen means it is essential to implement precautions and consider the risk factors of S. aureus infections in individuals with impaired immune systems, such as those with co-morbidities and chronic pathologies (patients with diabetes, heart disease, or chronic obstructive pulmonary disease (COPD)), those on dialysis, and those undergoing surgery [7,8,9,10]. In addition, it is not only necessary to understand S. aureus’s molecular and genetic factors that drive its virulence during the specific diseases it causes, but it is also important to strengthen our understanding of the factors that enable its colonisation, which is a major risk factor for these patients.
Further to its transit into various human tissues, S. aureus harbours numerous genetic loci that enable its resistance to antibiotic treatment. The development and transfer of these genes increases the complexity in treatment regimes. Combined with its dynamic metabolic range, specific virulence factors and the possession of these resistance genes underpin much of the chronic nature of S. aureus infections. However, it has become apparent in recent years that the failure of medical intervention in S. aureus infectious diseases, and thereby the basis for their relapse, is not limited to the possession of a specific set of virulence or antibiotic resistance genes but includes a broader shift in the bacterial cell types that permits a small number of quasi-dormant S. aureus cells to hide from the immune system and tolerate antimicrobial treatments. This review will present the current understanding of the development of these cell types during colonisation and extend the knowledge on antibiotic resistance to the phenotypic variation as a core attribute of subsequent chronic and relapsing infections.
2. Colonisation of Humans by Staphylococcus aureus
S. aureus has been found to have a distinct preference for specific areas during human colonisation, primarily the anterior nares, gastrointestinal tract, skin (most commonly the axillae and groin), throat, and vagina [11]. The anterior nares have long been established as the major reservoir for colonising S. aureus, with approximately 20–30% of the healthy population being permanent asymptomatic carriers and approximately 60–75% being intermittent carriers [4,12]. In contrast, healthy human skin is thought to offer unfavourable conditions for the establishment of permanent S. aureus skin colonisation. The basis for this remains unknown; however, it is speculated that the large surface area of the skin contributes to a lower density of colonising microbes that are constantly competing for regularly fluctuating nutrient availability and defects within the skin barrier [13]. It is also speculated that S. aureus is more likely to only temporarily colonise particular areas of the body, such as the skin, as a result of the bacteria hiding within or permanently colonising other niches of a single person. S. aureus’s successful colonisation of different sites around the human body is attributed to the specific molecular and genetic adaptations that allow it to adhere to its human host and evade therapeutic and host-generated stresses.
2.1. S. aureus and Mechanisms for Colonisation of Human Tissue
During nasal and skin colonisation, S. aureus has been found to highly express genes encoding for microbial surface components recognising adhesive matrix molecules (MSCRAMMs) (sdrC, sdrD, sasG, isdA, atlA, clfB, eap, fndA), cell surface dynamic and remodelling enzymes (atlA, sceD, oatA), wall teichoic acid (WTA) biosynthesis (tarK, tagO), and immune-modulatory factors (spa, chp, sap, sak) [14,15,16]. In contrast, S. aureus’s quorum-sensing accessory gene regulator (agr) system, which is responsible for the induction of protease and cytolytic toxin production and the downregulation of surface proteins, has been found to have reduced expression in colonising strains [16,17,18]. As a result, genes encoding for several toxins, including haemolysins (hla, hld, hlg), phenol-soluble modulins (psm), and bi-component leukotoxin homologue (blhB), are downregulated [17,18,19] (see Figure 1).
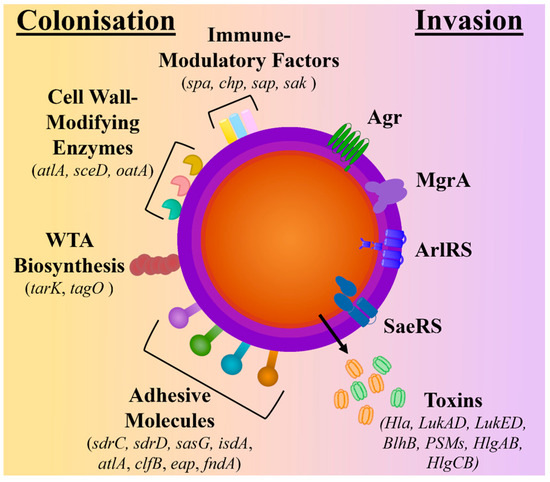
Figure 1.
Comparison of molecular factors upregulated during colonisation and invasion. The schematic depicts the surface proteins known to be upregulated during colonisation and includes adhesive molecules, wall teichoic acid (WTA) biosynthesis molecules, cell wall-modifying enzymes, and immune-modulatory factors. In contrast, during invasion, the regulatory systems (Agr quorum-sensing systems ArlRS, SaeRS, and MgrA) are upregulated, which controls the expression and secretion of toxins.
Unlike when colonising the nose, S. aureus within the skin microbiome must overcome the physical permeable barrier and chemical antimicrobial challenges that make up the human skin. An example of one of these stressors is the excretion of antibacterial fatty acids (AFAs) produced by sebaceous glands [20]. To overcome this, S. aureus over-expresses genes encoding for WTA and the iron-regulated surface determinant A (IsdA), which have both been shown to protect the bacteria from AFAs [20,21]. IsdA has also been shown to protect S. aureus from antimicrobial peptides (AMPs) in nasal secretions by binding to lactoferrin and subsequently inhibiting its proteolytic activity [22]. In addition, colonising S. aureus’s ability to express several adhesion proteins highlights the multi-functional nature of its interactions with its host. The abundance of different adhesive proteins presents a challenge when developing prophylactic treatments (such as vaccines) for decolonisation, as the selection of one or two proteins (e.g., ClfB or IsdA) could easily result in the bacteria employing other adhesins for colonisation [23]. A previous study analysing the gene expression of S. aureus during colonisation over 10 days showed variations in the expression of adhesive proteins at different stages of colonisation. Genes involved with WTA biosynthesis were found to be important during the early stages of colonisation, suggesting its importance for the initial bacteria–host interactions that bind to the scavenger receptor class F (SCRE-1) expressed by epithelial cells [24], whereas clfB and isdA were upregulated in the later stages, indicating their importance for maintaining attachment to the host tissue [18,19,25] by binding to iron-regulated surface determinant A (IsdA) [26], as well as for interactions with cytrokeratin and keratinised nasal cells (such as cytokeratin 10) [27] and the cell envelope protein loricrin [23].
This temporal regulation of genes during colonisation permits S. aureus to accumulate over time, allowing it to potentially regulate specific genes (such as via quorum-sensing and virulence pathways using Agr) and thereby switch to a pathogenic state when the host’s immune defences are compromised. This transit from colonisation to the infection site can be caused by aspiration, surgery, shaving, or catheter insertion [11,12,28,29]. These infections can therefore be caused by the bacteria already existing within the host (initially as commensals) and are referred to as autoinfections or endogenous infections. As such, individuals who are either persistent or intermittent carriers of S. aureus are at a higher risk of disease when their immune systems become compromised [29]. Additionally, it has been shown that polymorphisms within the host genome, such as interleukin-4 (IL-4), C-reactive protein (CRP), complement factor H (CFH) [30], defensins [31], mannose-binding lectin genes [32], and Toll-like receptors (TLRs) [32,33], frequently correlate to the colonisation of S. aureus, but there are only a small number of studies on the relation between host genes and S. aureus colonisation, leaving much nuance regarding the impact of these host genetic polymorphisms and the heritability of S. aureus colonisation [33].
2.2. Interactions between Colonising Staphylococcal Species
The human body harbours a rich array of densely populated microorganisms that interact and compete with one another through a range of mechanisms in order to uphold homeostasis and successfully colonise [34]. Different anatomical niches within the human host, including the nose, skin, and gastrointestinal tract, harbour unique microbiomes that differ in their phylogenetic and functional gene compositions [5]. Research efforts focusing on the nasal and skin microbiome have found that both environments are predominately populated by the Corynebacterium, Propionibacterium, Streptococcus, and Staphylococcus genera of bacteria [35]. From the colonising Staphylococcus species, the most commonly identified members include not only S. aureus but also the coagulase-negative Staphylococci (CoNS) species S. epidermidis, S. lugdunensis, S. capitis, S. warneri, S. haemolyticus, and S. hominis [36]. These CoNS species that are in direct competition with S. aureus have developed unique survival tactics to either inhibit or outright kill S. aureus without invading and causing infection to the host directly (Figure 2) [37]. These mechanisms include the biosynthesis of lantibiotics and bacteriocins. Recent studies have focussed on these bacterial compounds and their role in allowing for their dominance of local environments, such as the anterior nares and skin, specifically using these compounds also to target MRSA (methicillin-resistant S. aureus) and MDR (multi-drug-resistant) S. aureus.
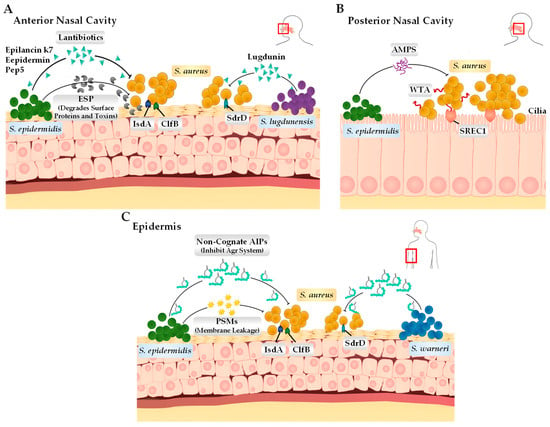
Figure 2.
Bacterial adhesin and the interactions between coagulase-negative Staphylococcus species and S. aureus on the human epithelium. (A) In the anterior nasal cavity, S. lugdunensis and S. epidermidis produce lantibiotics (S. lugdunensis produces lugdunin and S. epidermidis produces epilancin k7, epidermin, and Pep5) that can directly kill S. aureus. Additionally, S. epidermidis can also produce extracellular serine protease that cleaves surface proteins such as IsdA, ClfB, and SdrD, interfering with the S. aureus ability to adhere to the epithelial surface. (B) In the posterior nasal cavity, S. aureus binds to the scavenger receptor class F membrane 1 (SREC1) through interactions with wall teichoic acid (WTA) present on the cell surface. S. epidermidis expresses its antimicrobial peptides (antibiotics and ESP) to either kill or inhibit S. aureus cells. (C) On the skin surface (epidermis), S. warneri and S. epidermidis produce autoinducing peptides (AIPs) through their Agr systems, which is deemed important for survival specifically on the skin surface. S. epidermidis can also produce phenol-soluble modulins (PSMs), which can disrupt the membrane of S. aureus, causing leakage.
2.2.1. Staphylococcus epidermidis
Some strains of S. epidermidis have been found to synthesize and release extracellular serine protease (Esp). Esp has been found to degrade various proteins that are essential for adhesion to host surfaces and cohesive attachment to other bacteria for biofilm formation during colonisation [38]. The specific bacterial surface proteins degraded by Esp during nasal colonisation shown in in vitro and in vivo studies include adhesion molecules (SdrD, Emp, Eap, IsdA), cell surface dynamic and remodelling enzymes (Atl, SceD), immune-modulatory factors (Spa, Sbi), fibronectin-binding proteins (FnBPA, Efb), and β-haemolysin (Hlb). Esp was also found to degrade host receptor proteins, such as fibronectin, fibrinogen, and vitronectin, which are also important for the colonisation of S. aureus [39]. Through this action, Esp therefore acts indirectly for S. epidermidis to out-compete S. aureus within these host sites. Similar to interactions between Esp and S. aureus in the anterior nares, S. epidermidis has also evolved strategies to inhibit or kill other competing bacteria during skin colonisation. In particular, S. epidermidis expresses molecules that target colonising S. aureus. These include PSMs (PSM-γ and PSM-δ) that cause membrane leakage, cognate autoinducing peptides (AIPs) that disrupt quorum sensing by inhibiting the Agr system [40,41], and bacteriocins (Epidermin, Pep5, and Epilancin K7) that disrupt the barrier of the microbial cytoplasmic membrane [5].
2.2.2. Staphylococcus lugdunensis
S. lugdunensis is a prominent example of a CoNS species that generates a potent antimicrobial peptide and does not out-compete its bacterial neighbours. Specifically, it synthesises lugdunin, which has a bactericidal effect on colonising S. aureus [42]. Lugdunin is a lantibiotic that is synthesised by a Non-Ribosomal Peptide Synthase (NRPS) operon, which consists of four NRPS genes (lugA, B, C, D) [42]. In the anterior nares, lugdunin inhibits target microbes by dissipating their membrane potential, and it is theorised to achieve this using a protonophore-like mechanism [43]. Additionally, lugdunin is also capable of stimulating host skin cells to produce peptides that have antimicrobial properties and, when in synergy with lugdunin, can eliminate S. lugdunensis’s neighbouring and susceptible bacteria [44].
2.2.3. Staphylococcus warneri
The interactions between S. warneri and S. aureus have not been greatly researched. However, a recent study examined the mechanisms used by various colonising CoNS species to interfere with the quorum sensing of S. aureus through intra- or interspecies cross-talk. It was found that S. warneri secretes AIPs, specifically AIP-II, which has a potent inhibitory effect on the MRSA Agr system. S. warneri AIP-I still inhibited Agr but had a much weaker interaction with the S. aureus Agr system [45]. Coinfection experiments with the two bacteria also showed that S. warneri was capable of preventing transepithelial water loss and ultimately preventing barrier erythema and scaling, a strong indicator that S. warneri and its AIPs repress the Agr system of S. aureus and, consequently, the production of its virulence factors during invasion, and possibly also its colonisation of the skin. AIP-II was ultimately determined to be an effective inhibitor of MRSA agr-IV, with the capability of reducing the dermo-necrotic lesion size during skin infections [46]. It is important to take into account that there are no other studies that have investigated the interactions between S. aureus and S. warneri, or how S. warneri may play an important role in preventing or controlling S. aureus growth during colonisation and/or invasion. Thus, there is clearly still much nuance that remains unknown around the role that S. warneri plays as part of the CoNS commensal species and its subsequent antimicrobial effects on potentially pathogenic bacteria such as S. aureus.
2.2.4. Staphylococcus aureus Interactions with CoNS Species
In addition to the previously detailed CoNS species, S. aureus has also been found to have direct and dynamic interactions with other CoNS species that can inhibit or kill colonising S. aureus, such as S. capitis, S. chromogenes, S. pseudintermedius, and S. epidermidis, which have all been found to synthesise the purine analog 6-thioguanine (6-TG), a molecule found to inhibit agr quorum sensing, resulting in a reduction in the virulence and growth of S. aureus [47]. Furthermore, S. hominis [48], S. caprae [49], and S. simulans [50] have also been shown to produce novel, short autoinducing peptides that interfere with and ultimately block S. aureus quorum sensing, resulting in a reduction in bacterial growth [45,51].
S. capitis has also been found to produce a bacteriocinbiotic known as gallidermin, which can bind to the cell wall precursor lipid II of S. aureus and inhibit cell wall synthesis [52,53]. Alternatively, it has also been shown to inhibit WTA synthesis (which, as previously discussed, is important in the early stages of colonisation) [54]. S. hominis also secretes a bacteriocin known as micrococcin P1, which has been demonstrated to not only reduce S. aureus growth during infection but also to accelerate S. aureus-infected wound healing [55,56].
3. Changes in S. aureus Lifestyle during Colonisation
The variation in cell types within a single population of S. aureus cells during its different infections (such as OM and CF) have been a focus in numerous studies specifically directed at understanding chronic and relapsing infections. However, it is hypothesised that the presence of persistent cell types such as small colony variants (SCVs) and those that are predisposed to forming biofilms are not limited to S. aureus during a pathology but also exist within a population of colonising S. aureus. The ability to switch to a quasi-dormant cell type (or for such a cell type to exist as a subpopulation) provides multiple advantages in the overall survival of the bacteria, mainly via a slower metabolic rate. This consequently allows S. aureus to adapt to changes in different environmental stresses within the host, such as the presence of antibiotics, pH levels, competition with other colonising bacteria, and nutrient starvation stresses [57]. Unlike the growth rate of typical S. aureus, these quasi-dormant subpopulations are defined by their slow metabolic rate but prolonged survival under physical and chemical stresses, as well as their ability to live within (intra- and extracellular) host tissue, often without an immune response. Currently, the incidence of these subpopulations is well described for specific S. aureus infections; however, the presence of these cell types of S. aureus during colonisation is not well understood.
3.1. S. aureus and Biofilm during Colonisation
S. aureus’s ability to form complex biofilms is a contributing factor to the bacteria’s pathogenesis. Biofilms frequently form on the artificial surfaces of biomedical devices (such as medical implants and catheters) but have also been found to form on host tissues and during colonisation [58,59]. These biofilms are characterised by static, multiple layers of spatially grown heterogeneous single cells and microcolonies held together by a matrix referred to as an extracellular polymer substance (EPS) [60,61,62]. This matrix variously comprises proteins, extracellular DNA (eDNA), polysaccharide intercellular adhesin (PIA), and amyloid fibrils [60,62,63].
Colonisation, for example, of the skin, by S. aureus is initiated through adhesion by its surface-anchored proteins, such as SasG, to human cell types, particularly corneocytes. SasG (and other surface proteins that act as adhesins) also function in adhesion to nasal epithelial cells and are known to be functional in biofilm formation. Indeed, there are a large number of S. aureus adhesins that bind to human extracellular matrix (hECM) proteins and act as the beginning for biofilm formation and colonisation. However, S. aureus also has the ability to form planktonic aggregates, which are similar to biofilms but are distinct in composition and activity. Although biofilms are well characterised during invasion and are linked to persistent infections, their role in colonisation is not definitive. Currently, there is some evidence that biofilms are important in different sites for colonisation, but this seems to be transient, and there are contradictory reports that S. aureus forms biofilms during colonisation [5,37,64]. Dense bacterial populations (such as those in biofilms) have been shown to activate the Agr system (which, as previously discussed, is downregulated during colonisation), which allows for the expression of toxins and other proteins that favour an invasive phenotype [18,19]. Specific bacterial species are also commonly found to co-colonise human hosts with S. aureus, and these can secrete specific extracellular molecules that inhibit biofilm formation, such as the previously discussed ESP expressed by S. epidermidis [39,65,66,67].
Some studies have shown that S. aureus is dispersed and not consistent with a biofilm during nasal colonisation. Finally, because nasal-colonising S. aureus can be eradicated using mupirocin, it is presumed that biofilms do not form in the nasal cavity, as it would be likely that they would persist even after antimicrobial intervention [68]. However, there have been studies that have produced results showing that during colonisation, there are direct interactions between S. aureus and other bacteria that favour single-species S. aureus biofilms and promote polymicrobial biofilm formation, such as the co-colonisation of S. aureus and Propionibacterium spp. as a result of Propionibacterium-produced coproporphyrin III (CIII), which has been shown to induce biofilms and the coaggregation of S. aureus [69]. Due to the low number of bacteria, such as S. aureus, that populate the skin and nose, it is heavily suggested that S. aureus appears predominately as small clusters or dispersed cells [19].
3.2. S. aureus Small Colony Variants
SCVs are stochastically generated as a subpopulation within a normal population of actively growing cells. SCV cells exist within this population with a slow growth rate and an atypical colony morphology and have antibiotic tolerance as part of a population of antibiotic-susceptible cells [70]. Their phenotypic characteristics have been identified for over 100 years from clinical samples, with colonies observed to be slow-growing, non-pigmented, and approximately 1/10th the size of typical colonies (Figure 3). They also have reduced coagulase activity, reduced hemolysin production, increased antibiotic tolerance, and decreased membrane potential [71,72]. As a result of these characteristics, SCVs are often misdiagnosed as not S. aureus. The persistent phenotypic variation in SCVs has also been well described, such as their presence during S. aureus infections from soft tissue infections, bacteraemia, CF, OM, brain abscess, and device-related infections [71,73]. They have also been directly linked to unsuccessful treatment with long-term antibiotics [74], having been shown to tolerate a range of antibiotics, including fluoroquinolones, trimethoprim–sulfamethoxazole (SXT), fusidic acid, and aminoglycosides [75]. Although SCVs are well studied, they remain a clinical challenge to overcome when treating chronic S. aureus patients, or even in the development of a vaccine to decolonise S. aureus in healthy populations. SCV development is impacted, and formation is enhanced, by a variety of alternative metabolic pathways. These changes in metabolism have been related to various auxotrophisms, reduced ATP production, downregulated membrane potential, reduced CO2 biosynthesis, changes in the function of global regulatory proteins, reduced cell wall biosynthesis, and reduced fatty acid biosynthesis (Figure 4) [71,76,77,78].

Figure 3.
Comparison of typical S. aureus colonies, non-pigmented S. aureus colonies, and small colony variant (SCV) colonies grown on trypticase soya agar (TSA). Typical S. aureus produces large colonies with golden pigmentation, and in order to reduce the fitness cost, some strains of S. aureus present as large, non-pigmented colonies, whilst SCVs produce small, pinpoint colonies that are non-pigmented.
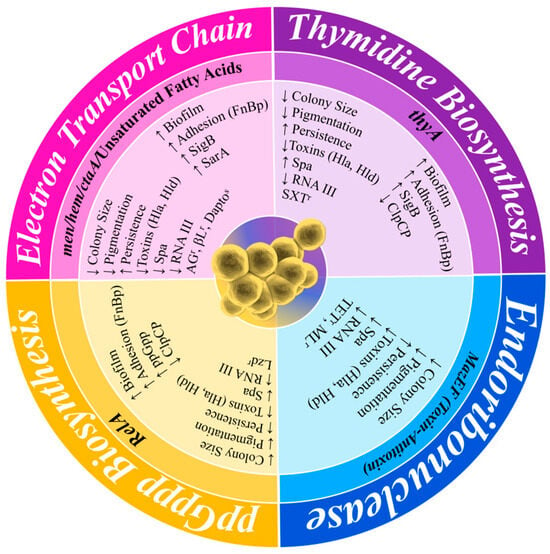
Figure 4.
Comparison of the phenotype–genotype variation in SCVs resulting from different auxotrophic mutations. The outer layer of the figure shows the phenotypical change, the central layer shows the auxotrophic mutations, and the inner layer shows the phenotype of each mutation or specific gene and the increase and decrease in activity. Largely, this is displayed by the differences in their auxotrophic mutations. Hla, α-toxin; Hld, δ-toxin; Spa, Protein A; SigB, σ-factor B; SarA, Staphylococcal accessory regulator A; FnBP, fibronectin-binding protein; ClpCP, ATP-dependent protease C/P; AGr, aminoglycoside resistance; βLr, beta-lactam resistance; Daptos, daptomycin susceptibility; TETr, tetracycline resistance; MLr, macrolide resistance.
Further complicating studies and clinical practice, there are different versions of SCVs. Electron transport-defective SCVs have a decreased biosynthesis of two essential components involved in the electron transport chain: menadione, essential in the biosynthesis of menaquinone caused by a mutation in menD and then in hemB (haemin), a porphyrin used in cytochrome biosynthesis [79,80]. Other mutations that affect electron transport include a mutation in ctaA, inhibiting haem A biosynthesis, which is also important for cytochrome biosynthesis [57,81]. Whilst SCVs with mutations in menadione and haemin have been isolated in patients suffering from infection, especially whilst undergoing treatment with aminoglycosides [74], the phenotypic variation displayed by these SCVs has been shown to be reversible through supplementation with the defective element, thereby supporting the clinical relevance of defects in the biosynthetic pathway of menadione and haemin [71,78,82]. Studies into hemB mutants in S. aureus SCVs have also shown that these have a reduction in α-toxin production [83], as well as the increased expression of clfA and fibronectin-binding protein (fnb) genes [84,85]. The increase in clfA and fnb allows for the mutant SCVs to have a greater ability to bind to the fibrinogen and the fibronectin of the host cells [84,85,86]. The ctaA mutant found in some SCVs has been shown to produce phenotypic variation within the colonies not only by a reduction in the colony size, but also by a decrease in the toxin production of α-toxin and toxic-shock-syndrome toxin 1 (TSST 1), a decrease in the expression of RNAIII, and an increase in resistance to aminoglycoside antibiotics [81].
Thymidine biosynthesis-defective SCVs are primarily related to patients with CF and patients undergoing long-term treatment with SXT [87]. The production of these SCVs is stimulated by SXT, as it interferes with the synthesis of tetrahydrofolic acid, an enzyme essential in the synthesis of thymidylate synthase encoded by the thyA gene. Unlike typical SCVs, thymidine-dependent SCVs can also exhibit two different phenotypical growth characteristics: the typical pinpoint colonies and colonies with an elevated, pigmented centre surrounded by a translucent edge on Columbia blood agar. These are referred to as “fried egg” colonies [88]. These phenotypes are also reversible by supplementation with thymidine [89].
In addition to auxotrophic SCVs, the S. aureus MazEF toxin–antitoxin system is also suspected to be involved in the generation of SCVs. This toxin–antitoxin system is transcribed as an operon where MazF (toxin) is readily bound to MazE (antitoxin), thereby neutralising the self-harming toxicity of MazF [57]. The antitoxin MazE is an RNase that interacts with hla, spa, and sigB mRNAs whilst avoiding sarA, recA, and gyrB mRNAs [90]. Increased production of MazF (beyond the capacity of MazE) gives rise to the typical SCV phenotype with a decreased growth rate and pigmentation with an increased survivability under various stresses [91]. Similarly, with an increase in the ATP-dependent protease system, ClpCP targets and reduces the MazE levels, allowing for an increase in MazF [70,92].
SCVs have increased expression of downregulators of the accessory gene regulator (agr) pathway, such as SrrAB, CodY, SigB, PSMs, and ArIRS. Meanwhile, positive regulators of Agr exhibit either reduced expression (SarA) or their expression is inhibited (MgrA) [75,93,94,95,96,97,98]. These changes in the regulation of the Agr operon ultimately result in a reduction in RNAIII production and therefore the inhibition of toxin expression, such as hla and TSST-1 [99,100]. However, there are exceptions to this pattern, with the SarA expression being downregulated in thymidine-dependant SCVs whilst being upregulated in hemB and menD mutants [101].
There are other genetic changes found to be associated with different SCVs, including mutations in relA, which encodes for RelA hydrolase. A reduction in the activity of RelA hydrolase results in the accumulation of the guanosine 3′,5′-bis(diphosphate) (ppGpp), an intracellular signalling molecule that causes the permanent activation of the stringent response [102]. Other enzymes that increase the synthesis of ppGpp are also dysregulated by Rsh, RelP, and RelQ [94]. The permanent activation of the stringent response limits protein synthesis, thereby reducing cellular growth and producing small colonies, and it is directly linked to linezolid resistance [102,103].
3.3. Selection for Small Colony Variants within the Polymicrobial Microbiome
An important developing area of understanding is the nature of polymicrobial niches that influence the phenotypic variation within S. aureus populations, such as those observed in patients with CF [104,105]. These studies have observed that signal molecules released by Pseudomonas aeruginosa, such as 4-hydroxyl-2-heptyl quinoline-N-oxide (HQNO), hydrogen cyanide, and pyocyanin, can protect S. aureus against aminoglycosides [106]. P. aeruginosa releases these molecules as secondary metabolites and these act as respiratory inhibitors of the growth of S. aureus by inhibiting S. aureus electron transport [104,106]. This does not impact SCVs (already not using the electron transport chain) and creates favourable conditions for SCV cells to continue to survive and thereby dominate within a population of S. aureus cells.
Within an anatomical niche, the constant battle for survival between colonising bacterial species creates different stresses that can be detrimental to the success of actively growing S. aureus. As previously established in this review, CoNS such as S. epidermidis, S. lugdunensis, and S. warneri can produce exoproducts, such as bacteriocins, proteases, AIPs, and PSMs, that can inhibit or kill S. aureus.
In addition to the stresses from CoNS species, Streptococcus pneumoniae is able to kill S. aureus due to its production of hydrogen peroxide [107,108,109]. Hydrogen peroxide causes irreversible damage to specific protein groups by oxidising iron groups or product OH-, resulting in DNA damage. Much like the selective pressure from HQNO from P. aeruginosa, hydrogen peroxide has also been shown to select for aminoglycoside-resistant SCVs that have mutations predominantly in menD and less so in hemB [110]. It has also been shown that hydrogen peroxide, when used as a method of decolonisation, is unsuccessful at eradicating S. aureus in the nasal cavities of neonatal rats [111].
Cornyebacterium spp., like S. aureus, has also been well established as a commensal within the nasal cavity [35]. Cornyebacterium spp. (such as C. pseudodiphtheriticum) have been shown to inhibit the S. aureus Agr system suspected to be the result of the expression of inhibitory AIPs or PSMs [112]. As has already been well established, S. aureus downregulates agr in order to favour colonisation factors, biofilms, and SCVs [113,114], and the inhibition of the agr system from other bacterial species can be problematic to individuals who are immunocompromised. Alternatively, Cornyebacterium spp. can directly kill S. aureus and therefore potentially drive it to turn off its virulence gene expression in favour of a more persistent lifestyle (SCV) and to escape being killed [112].
4. Antibiotic Responses of S. aureus Lifestyles
4.1. Antibiotic Resistance by S. aureus Strains
A wide range of S. aureus species have evolved over time to develop specific mechanisms that allow them to survive and continue growing in the presence of different antibiotics [104]. These antibiotic-resistant strains of S. aureus have become highly prevalent, especially in hospital-acquired infections. The most significant strains with an impact on both hospital- and community-acquired bacterial infections are the MRSA strains [115]. Methicillin, like penicillin, is a β-lactam class of antibiotics, and resistance occurs through the acquisition of the mecA gene located on the Staphylococcal Chromosomal Cassette (SCCmec). This SCC element is a large fragment of DNA that is categorized as a mobile genetic element (MGE) and often encodes antibiotic resistance and/or virulence determinants that can be passed on between S. aureus populations through horizontal gene transfer [115,116]. As well as MRSA, another common antibiotic-resistant S. aureus population is vancomycin-resistant S. aureus (VRSA). Vancomycin is a glycopeptide class of antibiotic for which resistance arises when the developing mutations in the vanA operon, which consists of the vanRSHAXYZ genes, cause modifications to the cell wall that prevent vancomycin from binding efficiently [117,118]. S. aureus has also been found to develop resistance to other antibiotic classes, including but not limited to aminoglycosides, tetracycline, oxazolidinone, macrolides, fluoroquinolones, rifampicin, and lipopeptides [119,120,121], summarised in Table 1. An equal concern is the growing population of the different multi-drug-resistant (MDR) strains of S. aureus that have complete resistance to three or more antibiotic classes, for example, S. aureus strains that are resistant to oxacillin (β-lactam), ciprofloxacin (fluoroquinolones), linezolid (oxazolidinone), and erythromycin (macrolide) [122].

Table 1.
Antimicrobial resistance genes and mechanisms in Staphylococcus aureus.
4.2. Antibiotic Tolerance: Indirect and Direct Responses by S. aureus to Antibiotics
In contrast to antibiotic resistance, there is the ability of a bacterial strain to temporarily withstand or slow down the lethal consequences of high doses of antibiotics, measured by the decrease in antibiotic killing and resulting in continued bacterial survival [123]. These tolerant bacterial strains are typically genetically susceptible to antibiotic killing [124], unlike antibiotic resistance strains that have mutations in specific genes that allow them to resist antibiotic treatment by a broad shift in the bacterial phenotype. Tolerance can arise from a stochastic change in metabolic functions, causing a reduction in antibiotic uptake and preventing the killing of various antibiotic classes [125]. This phenotype is thought to present itself as either tolerance by “slow growth” (occurring in a steady state) or “by lag” (an extended lag phase of growth, which is a transient state as a response to stress conditions) [126]. An example of an S. aureus phenotype that exhibits antibiotic tolerance is the SCV phenotype, which, as previously mentioned, exhibits mutations that favour slow growth and changes in the metabolic pathways that allow for antibiotic tolerance [127]. The antibiotic targets are almost always the molecular mechanism during growth, and in a slow or no-growth state, these targets are not functional. It is currently understood that antibiotic tolerance is a phenomenon synonymous with persister cell phenotypes; however, there is no known identifying genetic marker that can be directly linked to these cells’ ability to tolerate antibiotics [128]. Thus, due to the difficulty in isolating persister cells, and SCVs, from clinical samples, these subpopulations that are tolerant to antibiotic treatments can exist within a population of cells that display antibiotic susceptibility, and can persist and ultimately result in chronic or relapsing infections [129].
4.3. Heteroresistance and S. aureus
Defining antibiotic resistance has been a strong focus in the research for the past decades, focusing on mutations and the acquisition of various genetic changes that differ from the typical to the genes in typical susceptible phenotypes [130]. However, there are exceptions to the correlation between the presence of resistant genes and the observed resistant phenotypes. An example of this exception is the phenomenon of heteroresistance, where subpopulations present within a heterogeneous cell population, possessing cells with elevated levels of resistance to antibiotics compared to those of the dominant cell population [131]. Unlike antibiotic-tolerant bacterial strains, heteroresistant populations proliferate under antibiotic stress and can arise either intrinsically (without pre-exposure to antibiotics) or via acquisition (from initial exposure to antibiotics) [132]. This ultimately gives rise to subpopulations of persistent cell types, such as those that are predisposed to form biofilms or even SCVs [133]. The low population of these heteroresistant strains within a single isolate makes it increasingly difficult to reliably determine an accurate susceptibility profile of the bacterial strain. Currently, the methods to determine the presence of heteroresistant strains include population analysis profiling (PAP), Etest, disc diffusion, and agar dilution [134,135]. Despite these established assays, heteroresistant strains remain easy to miss and are suggested to play an important role in prolonged infections and mortality rates [136]. This has become more frequently observed in vancomycin-intermediate S. aureus (heteroresistant VISA (hVISA), which includes intermediate strains but with subpopulations that display resistance) within hospital settings [117].
The current understanding is that heteroresistance results from a few different mechanisms can be categorised as either stable (where resistance persists after the removal of the antibiotic stress) or unstable (where the population reverts back to an antibiotic susceptible phenotype after the antibiotic selection pressure has been removed) [135] (Figure 5). Stable heteroresistance is frequently observed to be a result of mutations that are typically genetically stable, such as single-nucleotide polymorphisms (SNPs), frameshifts, insertions, and deletions [135]. These mutations generally have a marginal impact on the fitness cost, allowing the subpopulation to remain resistant to specific antibiotics even once the stress has been removed. These stable phenotypes have been observed in both Gram-positive bacteria such as S. aureus (e.g., hVISA) and Gram-negative bacteria such as Acinetobacter baumannii [134]. Alternatively, unstable heteroresistance is thought to be a result of spontaneous, genetic, tandem amplification, where an increase in resistance genes present within the bacterial genome gives rise to increased resistance [137]. This unstable phenotype has been almost exclusively observed in Gram-negative bacterial species, such as Escherichia coli and Haemophilus influenzae, due to the spontaneous homologous recombination between repeated sequences in sister chromatids during replications [138]. Thus, amplified gene regions increase resistance corresponding to an increase in the gene dosage (such as genes encoding for antibiotic resistance, efflux pumps, or antibiotic-modifying/-degrading enzymes) [139]. However, due to the high fitness cost of amplification, the gene is de-amplified when the stress is removed [140].
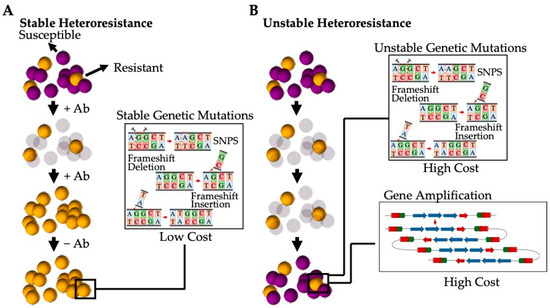
Figure 5.
Heteroresistance generated by a subpopulation of bacterial cells present as a result of varying genetic mutations. (A) Stable heteroresistant mutations are a result of genetic mutations that have low or no fitness cost and that favour a stable phenotype that is resistant to varying antibiotics. These mutations are typically the result of SNPs, frameshifts, and insertions and/or deletions. (B) Unstable heteroresistant mutations are the result of genetic mutations that have a high fitness cost and that revert to the original antibiotic susceptible phenotype when the selective pressure of the antibiotic is removed. These mutations are typically a result of spontaneous, tandem amplification or high-cost SNPs, frameshifts, and insertions and/or deletions.
A recent study looked at the genetic changes in heteroresistant S. aureus isolates compared to their parental strains. The prevalence of heteroresistance was studied in 40 parental clinical S. aureus isolates when grown in the presence of daptomycin, gentamycin, linezolid, oxacillin, teicoplanin, and vancomycin [141]. Amongst the 40 isolates, 52.5% exhibited heteroresistance to at least one of the antibiotics tested, 17.5% exhibited heteroresistance towards two, and 15% exhibited heteroresistance towards three, with only 15% of isolates exhibiting no heteroresistance to any of the antibiotics. Genetic mutations in each heteroresistant strain isolated were dependent on which antibiotic to which it displayed resistance. Mutations in known antibiotic-resistant genes were most commonly observed as a result of SNPs, frameshifts, insertions, or deletions typical to those seen in stable heteroresistance; however, mutations in genes associated with the heme and menaquinone pathways, transfer RNA (tRNA) modification, tRNA aminoacylation, and the ribosomes were also observed [141]. This study, although small, did illustrate the prevalence of heteroresistance in S. aureus, and that the regular mutations within the bacterial chromosome in different core genes are the main mechanisms by which heteroresistance is conferred. They also highlighted that the reason that Gram-positive bacteria, such as S. aureus, do not present with random gene amplification is due to the lack of resistance genes and repeat sequences present within the bacterial genome [141].
4.4. Clinical Significance of S. aureus Small Colony Variants and Colonisation
There are different and specific contexts to the significance of the variations in S. aureus cell types. S. aureus is one of the most common causative pathogens of infections worldwide, responsible for a vast array of nosocomial and community infections [142]. Chronic and relapsing infections have been linked to the presence of SCVs and the inability of antibiotics and the immune system to completely remove these cells. However, healthy individuals colonised with S. aureus have been shown to have an increased risk of invasive and non-invasive infections [68,143,144,145], with over 80% of S. aureus infectious strains found to be genetically similar and endogenous to those isolated from the nares of colonised individuals [6,146,147,148].
S. aureus is the most isolated pathogen from diabetic foot infections (DFIs), commonly resulting in persistent OM infections or DFI-OM [149]. It has recently been shown that ~39% (n = 109/276) of DFI patients were nasal carriers of S. aureus, whilst ~36% (n = 101/276) harboured S. aureus in both their nares and sites of infection. Of these patients, ~65% of S. aureus strains were genetically identical in both locations [150]. This has also been seen in previous studies looking at the relationship between nasal and diabetic foot ulcer (DFU) S. aureus colonies [104,147,151,152,153]. SCVs have also been recovered from DFI-OM patients [74]; however, the role SCVs have in persistent and relapsing infection is still not well studied.
In contrast to the unknown role of SCVs in DFI-OM by S. aureus, SCVs have been extensively studied in CF patients, of which approximately two-thirds are infected with S. aureus SCVs in conjunction with P. aeruginosa [154,155]. As previously mentioned, coinfection with P. aeruginosa and long-term aminoglycoside or SXT treatments creates environmental stresses within the host that select for the SCV phenotype within a population of S. aureus [104,156,157,158,159]. SCVs have been directly linked to poor clinical outcomes in CF patients, as they contribute directly to the chronic inflammation of the lungs [104,160]. Although the involvement of SCVs in CF patients has been well researched, the role that colonisation has in contributing to CF infections is not well characterised. This also holds true for device-related (such as prosthetic joints) infections and skin and soft tissue infections, for which the role of SCVs is well researched but the involvement of colonisation in persistence and relapse is not [161].
4.5. Diagnosis and S. aureus Small Colony Variants
A range of rapid non-molecular and molecular diagnostic methods are available to identify and diagnose S. aureus directly from clinical samples. There are selective and differential media that have been developed to either isolate or select for S. aureus by specifically taking advantage of the unique S. aureus enterotoxin and enzymes produced by the bacteria. Blood agar is commonly used as a differential media to identify haemolytic characteristics of bacteria that are also typical of S. aureus. However, the use of the selective media: Staphylococcal medium 110, Brilliance Blue, and Mannitol Salt Agar (MSA), allow for the isolated growth of S. aureus with minimal to no growth of E. coli over a 16–24 h incubation. Positive colonies can undergo further identification with a PBP2a latex agglutination assay, polymerase chain reaction (PCR) of the mecA gene, and susceptibility testing to determine antibiotic resistance [162]. More modern diagnostic techniques include culturing S. aureus on the selective medium chromogenic S. aureus ID agar, where S. aureus strains such as methicillin-resistant S. aureus (MRSA) can be detected based on the colour of the colonies present. Finally, S. aureus can be identified within 1–4 h through PCR alone from a collected nasal swab [162,163].
However, none of these methods take into account the cells within an S. aureus infection, which possess a slow growth rate or very low to no expression of the usual traits, as seen in SCVs. These cells can therefore remain undetected, making standardised testing methods difficult, and SCV-based infections or infections, including SCV cells as part of the infection, are often missed during the clinical identification of S. aureus [164]. These atypical S. aureus phenotypes are identified as non-haemolytic, non-pigmented, pinpoint colonies due to deficiencies or reductions in biochemical reactions [165]. Figure 3 provides a comparison of typical golden S. aureus growth, atypical non-pigmented active cell growth, and SCVs on tryptone soy agar (TSA). This includes a reduction in coagulase production, inhibiting their ability to grow on the traditional selective media Staphylococcal Medium 110, MSA, and Brilliance Blue [71,166]. Thus, the atypical morphologies and physiologies of SCVs present a challenge in the accurate clinical diagnosis of S. aureus. To avoid misidentification, extended conventional culturing and identification techniques must be implemented for the successful isolation of SCVs. Currently, culturing specimens on both blood agar and chromogenic S. aureus ID agar over 24–72 h incubation is the most rapid and accurate method of identifying S. aureus and their SCV phenotypes. Of note, SCVs have a growth rate of approximately one-ninth of typical S. aureus, causing them to be out-competed during the log growth phase [166]. To overcome this, isolates suspected of being S. aureus SCVs can undergo further confirmation through the use of molecular methods such as 16S rRNA partial sequencing [162], the amplification of species-specific DNA targets (coagulase, nuclease, mecA gene, nuc gene) [163], or an anti-Penicillin Binding Protein 2a (PBP2a) latex agglutination test [167]. However, the identification of SCVs is currently still a long process in comparison to the rapid diagnostics of typical S. aureus cell growth.
5. Future Directions and Diagnosis of S. aureus Lifestyles
S. aureus’s ability to switch to an alternative, more persistent cell type has been demonstrated to be an advantageous adaptation to alterations in the bacteria’s environment with persistent physical and chemical stresses. Previous studies have had a strong focus on different environmental stresses ranging from changes in pH, temperature, antibiotics, and a range of metabolic stresses. These changes within the host conditions cause the usually dominant, active S. aureus cells in a population to decrease in favour of a slower-growing, quasi-dormant phenotype, such as an SCV. The majority of these studies have focussed on characterising the changes in specific pathways as a result of mutations that alter the bacterial cell metabolism.
However, these studies are largely limited to S. aureus variations in cell types during infections, with very little analysis on these persistent SCV cell types or the proclivity for an active cell to switch to an SCV during the innocuous colonisation of healthy individuals. We postulate that some strains of S. aureus that asymptomatically colonise the different anatomical niches in the human host have the tendency to (1) grow as a single heterogenous population of cells that are made up of active cells, SCVs, and those predisposed to forming biofilms, and/or (2) have the ability to switch between the different alternative cell types during stress conditions.
In addition to this, the microbiomes of the various environmental niches within the human host exist as vast arrays of microorganisms that are constantly competing to establish themselves. The interactions between the different Staphylococci species have become a large focus in recent studies as part of the quest to identify molecules, such as bacteriocins, that can be utilised in place of or in conjunction with antimicrobial therapeutics as a potential strategy to combat the antibiotic resistance crisis.
Author Contributions
Conceptualization, C.M.B.-G. and S.P.K.; writing—original draft preparation, C.M.B.-G.; writing—review and editing, S.P.K.; supervision, S.P.K.; project administration, S.P.K.; funding acquisition, C.M.B.-G. and S.P.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Diabetes SA (South Australia), grant number 85381.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the members of the Kidd laboratory for their useful comments.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- GBD 2019 Antimicrobial Resistance Collaborators. Global mortality associated with 33 bacterial pathogens in 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef]
- Kwiecinski, J.M.; Horswill, A.R. Staphylococcus aureus bloodstream infections: Pathogenesis and regulatory mechanisms. Curr. Opin. Microbiol. 2020, 53, 51–60. [Google Scholar] [CrossRef]
- Sollid, J.U.; Furberg, A.S.; Hanssen, A.M.; Johannessen, M. Staphylococcus aureus: Determinants of human carriage. Infect. Genet. Evol. 2014, 21, 531–541. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Krismer, B.; Weidenmaier, C.; Zipperer, A.; Peschel, A. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat. Rev. Microbiol. 2017, 15, 675–687. [Google Scholar] [CrossRef] [PubMed]
- von Eiff, C.; Becker, K.; Machka, K.; Stammer, H.; Peters, G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N. Engl. J. Med. 2001, 344, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Munoz, P.; Hortal, J.; Giannella, M.; Barrio, J.M.; Rodriguez-Creixems, M.; Perez, M.J.; Rincon, C.; Bouza, E. Nasal carriage of S. aureus increases the risk of surgical site infection after major heart surgery. J. Hosp. Infect. 2008, 68, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Paling, F.P.; Hazard, D.; Bonten, M.J.M.; Goossens, H.; Jafri, H.S.; Malhotra-Kumar, S.; Sifakis, F.; Weber, S.; Kluytmans, J.; Team, A.-I.S. Association of Staphylococcus aureus Colonization and Pneumonia in the Intensive Care Unit. JAMA Netw. Open 2020, 3, e2012741. [Google Scholar] [CrossRef]
- Pongbangli, N.; Oniem, N.; Chaiwarith, R.; Nantsupawat, T.; Phrommintikul, A.; Wongcharoen, W. Prevalence of Staphylococcus aureus nasal carriage and surgical site infection rate among patients undergoing elective cardiac surgery. Int. J. Infect. Dis. 2021, 106, 409–414. [Google Scholar] [CrossRef]
- Sakr, A.; Bregeon, F.; Rolain, J.M.; Blin, O. Staphylococcus aureus nasal decolonization strategies: A review. Expert. Rev. Anti-Infect. Ther. 2019, 17, 327–340. [Google Scholar] [CrossRef]
- Gordon, R.J.; Lowy, F.D. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 2008, 46 (Suppl. S5), S350–S359. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, H.F.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Bier, K.; Schittek, B. Beneficial effects of coagulase-negative Staphylococci on Staphylococcus aureus skin colonization. Exp. Dermatol. 2021, 30, 1442–1452. [Google Scholar] [CrossRef]
- Howden, B.P.; Giulieri, S.G.; Wong Fok Lung, T.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.H.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 2023, 21, 380–395. [Google Scholar] [CrossRef]
- Edwards, A.M.; Massey, R.C.; Clarke, S.R. Molecular mechanisms of Staphylococcus aureus nasopharyngeal colonization. Mol. Oral Microbiol. 2012, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Burian, M.; Plange, J.; Schmitt, L.; Kaschke, A.; Marquardt, Y.; Huth, L.; Baron, J.M.; Hornef, M.W.; Wolz, C.; Yazdi, A.S. Adaptation of Staphylococcus aureus to the Human Skin Environment Identified Using an ex vivo Tissue Model. Front. Microbiol. 2021, 12, 728989. [Google Scholar] [CrossRef]
- Acker, K.P.; Wong Fok Lung, T.; West, E.; Craft, J.; Narechania, A.; Smith, H.; O’Brien, K.; Moustafa, A.M.; Lauren, C.; Planet, P.J.; et al. Strains of Staphylococcus aureus that Colonize and Infect Skin Harbor Mutations in Metabolic Genes. iScience 2019, 19, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Burian, M.; Wolz, C.; Goerke, C. Regulatory adaptation of Staphylococcus aureus during nasal colonization of humans. PLoS ONE 2010, 5, e10040. [Google Scholar] [CrossRef]
- Burian, M.; Rautenberg, M.; Kohler, T.; Fritz, M.; Krismer, B.; Unger, C.; Hoffmann, W.H.; Peschel, A.; Wolz, C.; Goerke, C. Temporal expression of adhesion factors and activity of global regulators during establishment of Staphylococcus aureus nasal colonization. J. Infect. Dis. 2010, 201, 1414–1421. [Google Scholar] [CrossRef]
- Kohler, T.; Weidenmaier, C.; Peschel, A. Wall teichoic acid protects Staphylococcus aureus against antimicrobial fatty acids from human skin. J. Bacteriol. 2009, 191, 4482–4484. [Google Scholar] [CrossRef]
- Clarke, S.R.; Mohamed, R.; Bian, L.; Routh, A.F.; Kokai-Kun, J.F.; Mond, J.J.; Tarkowski, A.; Foster, S.J. The Staphylococcus aureus surface protein IsdA mediates resistance to innate defenses of human skin. Cell Host Microbe 2007, 1, 199–212. [Google Scholar] [CrossRef]
- Clarke, S.R.; Foster, S.J. IsdA protects Staphylococcus aureus against the bactericidal protease activity of apolactoferrin. Infect. Immun. 2008, 76, 1518–1526. [Google Scholar] [CrossRef]
- Corrigan, R.M.; Miajlovic, H.; Foster, T.J. Surface proteins that promote adherence of Staphylococcus aureus to human desquamated nasal epithelial cells. BMC Microbiol. 2009, 9, 22. [Google Scholar] [CrossRef]
- Baur, S.; Rautenberg, M.; Faulstich, M.; Grau, T.; Severin, Y.; Unger, C.; Hoffmann, W.H.; Rudel, T.; Autenrieth, I.B.; Weidenmaier, C. A nasal epithelial receptor for Staphylococcus aureus WTA governs adhesion to epithelial cells and modulates nasal colonization. PLoS Pathog. 2014, 10, e1004089. [Google Scholar] [CrossRef] [PubMed]
- Weidenmaier, C.; Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef]
- Clarke, S.R.; Andre, G.; Walsh, E.J.; Dufrene, Y.F.; Foster, T.J.; Foster, S.J. Iron-regulated surface determinant protein A mediates adhesion of Staphylococcus aureus to human corneocyte envelope proteins. Infect. Immun. 2009, 77, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.M.; Walsh, E.J.; Massey, R.C.; Peacock, S.J.; Foster, T.J. Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: Implications for nasal colonization. Cell Microbiol. 2002, 4, 759–770. [Google Scholar] [CrossRef]
- Kluytmans, J.; van Belkum, A.; Verbrugh, H. Nasal carriage of Staphylococcus aureus: Epidemiology, underlying mechanisms, and associated risks. Clin. Microbiol. Rev. 1997, 10, 505–520. [Google Scholar] [CrossRef]
- Oliveira, D.; Borges, A.; Simoes, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Emonts, M.; Uitterlinden, A.G.; Nouwen, J.L.; Kardys, I.; Maat, M.P.; Melles, D.C.; Witteman, J.; Jong, P.T.; Verbrugh, H.A.; Hofman, A.; et al. Host polymorphisms in interleukin 4, complement factor H, and C-reactive protein associated with nasal carriage of Staphylococcus aureus and occurrence of boils. J. Infect. Dis. 2008, 197, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Nurjadi, D.; Herrmann, E.; Hinderberger, I.; Zanger, P. Impaired beta-defensin expression in human skin links DEFB1 promoter polymorphisms with persistent Staphylococcus aureus nasal carriage. J. Infect. Dis. 2013, 207, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Vuononvirta, J.; Toivonen, L.; Grondahl-Yli-Hannuksela, K.; Barkoff, A.M.; Lindholm, L.; Mertsola, J.; Peltola, V.; He, Q. Nasopharyngeal bacterial colonization and gene polymorphisms of mannose-binding lectin and toll-like receptors 2 and 4 in infants. PLoS ONE 2011, 6, e26198. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Rose, W.; Schrodi, S.J. Complex host genetic susceptibility to Staphylococcus aureus infections. Trends Microbiol. 2015, 23, 529–536. [Google Scholar] [CrossRef]
- Otto, M. Staphylococci in the human microbiome: The role of host and interbacterial interactions. Curr. Opin. Microbiol. 2020, 53, 71–77. [Google Scholar] [CrossRef]
- Sakr, A.; Bregeon, F.; Mege, J.L.; Rolain, J.M.; Blin, O. Staphylococcus aureus Nasal Colonization: An Update on Mechanisms, Epidemiology, Risk Factors, and Subsequent Infections. Front. Microbiol. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Kaspar, U.; Kriegeskorte, A.; Schubert, T.; Peters, G.; Rudack, C.; Pieper, D.H.; Wos-Oxley, M.; Becker, K. The culturome of the human nose habitats reveals individual bacterial fingerprint patterns. Environ. Microbiol. 2016, 18, 2130–2142. [Google Scholar] [CrossRef]
- Piewngam, P.; Otto, M. Staphylococcus aureus colonisation and strategies for decolonisation. Lancet Microbe 2024, 5, e606–e618. [Google Scholar] [CrossRef]
- Moon, J.L.; Banbula, A.; Oleksy, A.; Mayo, J.A.; Travis, J. Isolation and characterization of a highly specific serine endopeptidase from an oral strain of Staphylococcus epidermidis. Biol. Chem. 2001, 382, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Iwase, T.; Uehara, Y.; Shinji, H.; Tajima, A.; Seo, H.; Takada, K.; Agata, T.; Mizunoe, Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 2010, 465, 346–349. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcus aureus and Staphylococcus epidermidis peptide pheromones produced by the accessory gene regulator agr system. Peptides 2001, 22, 1603–1608. [Google Scholar] [CrossRef]
- Parlet, C.P.; Brown, M.M.; Horswill, A.R. Commensal Staphylococci Influence Staphylococcus aureus Skin Colonization and Disease. Trends Microbiol. 2019, 27, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Salazar, B.; Heilbronner, S.; Peschel, A.; Krismer, B. Secondary Metabolites Governing Microbiome Interaction of Staphylococcal Pathogens and Commensals. Microbial Physiol. 2021, 31, 1–19. [Google Scholar] [CrossRef]
- Bitschar, K.; Sauer, B.; Focken, J.; Dehmer, H.; Moos, S.; Konnerth, M.; Schilling, N.A.; Grond, S.; Kalbacher, H.; Kurschus, F.C.; et al. Lugdunin amplifies innate immune responses in the skin in synergy with host- and microbiota-derived factors. Nat. Commun. 2019, 10, 2730. [Google Scholar] [CrossRef]
- Williams, P.; Hill, P.; Bonev, B.; Chan, W.C. Quorum-sensing, intra- and inter-species competition in the staphylococci. Microbiology 2023, 169. [Google Scholar] [CrossRef]
- Severn, M.M.; Cho, Y.K.; Manzer, H.S.; Bunch, Z.L.; Shahbandi, A.; Todd, D.A.; Cech, N.B.; Horswill, A.R. The Commensal Staphylococcus warneri Makes Peptide Inhibitors of MRSA Quorum Sensing that Protect Skin from Atopic or Necrotic Damage. J. Invest. Dermatol. 2022, 142, 3349–3352.e5. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Goncheva, M.I.; Flannagan, R.S.; Deecker, S.R.; Guariglia-Oropeza, V.; Ensminger, A.W.; Heinrichs, D.E. Coagulase-negative staphylococci release a purine analog that inhibits Staphylococcus aureus virulence. Nat. Commun. 2021, 12, 1887. [Google Scholar] [CrossRef]
- Severn, M.M.; Williams, M.R.; Shahbandi, A.; Bunch, Z.L.; Lyon, L.M.; Nguyen, A.; Zaramela, L.S.; Todd, D.A.; Zengler, K.; Cech, N.B.; et al. The Ubiquitous Human Skin Commensal Staphylococcus hominis Protects against Opportunistic Pathogens. mBio 2022, 13, e0093022. [Google Scholar] [CrossRef]
- d’Ersu, J.; Aubin, G.G.; Mercier, P.; Nicollet, P.; Bemer, P.; Corvec, S. Characterization of Staphylococcus caprae Clinical Isolates Involved in Human Bone and Joint Infections, Compared with Goat Mastitis Isolates. J. Clin. Microbiol. 2016, 54, 106–113. [Google Scholar] [CrossRef]
- Peng, P.; Baldry, M.; Gless, B.H.; Bojer, M.S.; Espinosa-Gongora, C.; Baig, S.J.; Andersen, P.S.; Olsen, C.A.; Ingmer, H. Effect of Co-inhabiting Coagulase Negative Staphylococci on S. aureus agr Quorum Sensing, Host Factor Binding, and Biofilm Formation. Front. Microbiol. 2019, 10, 2212. [Google Scholar] [CrossRef]
- Paharik, A.E.; Parlet, C.P.; Chung, N.; Todd, D.A.; Rodriguez, E.I.; Van Dyke, M.J.; Cech, N.B.; Horswill, A.R. Coagulase-Negative Staphylococcal Strain Prevents Staphylococcus aureus Colonization and Skin Infection by Blocking Quorum Sensing. Cell Host Microbe 2017, 22, 746–756. [Google Scholar] [CrossRef]
- Szekat, C.; Josten, M.; Rickmeyer, J.; Crusemann, M.; Bierbaum, G. A Staphylococcus capitis strain with unusual bacteriocin production. Microb. Biotechnol. 2023, 16, 2181–2193. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Jangir, P.K.; Das, J.; Taneja, B.; Sharma, R. Genome Analysis of Staphylococcus capitis TE8 Reveals Repertoire of Antimicrobial Peptides and Adaptation Strategies for Growth on Human Skin. Sci. Rep. 2017, 7, 10447. [Google Scholar] [CrossRef]
- Muller, A.; Ulm, H.; Reder-Christ, K.; Sahl, H.G.; Schneider, T. Interaction of type A lantibiotics with undecaprenol-bound cell envelope precursors. Microb. Drug Resist. 2012, 18, 261–270. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Du, Z.; Zhang, L.; Chen, J.; Shen, Z.; Liu, Q.; Qin, J.; Lv, H.; Wang, H.; et al. Skin microbiota analysis-inspired development of novel anti-infectives. Microbiome 2020, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Demessant-Flavigny, A.L.; Connetable, S.; Kerob, D.; Moreau, M.; Aguilar, L.; Wollenberg, A. Skin microbiome dysbiosis and the role of Staphylococcus aureus in atopic dermatitis in adults and children: A narrative review. J. Eur. Acad. Dermatol. Venereol. 2023, 37 (Suppl. S5), 3–17. [Google Scholar] [CrossRef]
- Proctor, R.A.; Kriegeskorte, A.; Kahl, B.C.; Becker, K.; Loffler, B.; Peters, G. Staphylococcus aureus Small Colony Variants (SCVs): A road map for the metabolic pathways involved in persistent infections. Front. Cell Infect. Microbiol. 2014, 4, 99. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Berends, E.T.M.; Zheng, X.; Hill, P.J.; Chan, R.; Torres, V.J.; Wozniak, D.J. Leukocidins and the Nuclease Nuc Prevent Neutrophil-Mediated Killing of Staphylococcus aureus Biofilms. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Schilcher, K.; Horswill, A.R. Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol. Mol. Biol. Rev. 2020, 84. [Google Scholar] [CrossRef]
- Gotz, F. Staphylococcus and biofilms. Mol. Microbiol. 2002, 43, 1367–1378. [Google Scholar] [CrossRef]
- Trizna, E.Y.; Yarullina, M.N.; Baidamshina, D.R.; Mironova, A.V.; Akhatova, F.S.; Rozhina, E.V.; Fakhrullin, R.F.; Khabibrakhmanova, A.M.; Kurbangalieva, A.R.; Bogachev, M.I.; et al. Bidirectional alterations in antibiotics susceptibility in Staphylococcus aureus-Pseudomonas aeruginosa dual-species biofilm. Sci. Rep. 2020, 10, 14849. [Google Scholar] [CrossRef] [PubMed]
- Tuon, F.F.; Suss, P.H.; Telles, J.P.; Dantas, L.R.; Borges, N.H.; Ribeiro, V.S.T. Antimicrobial Treatment of Staphylococcus aureus Biofilms. Antibiotics 2023, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Ravaioli, S.; Montanaro, L. Polysaccharide intercellular adhesin in biofilm: Structural and regulatory aspects. Front. Cell Infect. Microbiol. 2015, 5, 7. [Google Scholar] [CrossRef]
- Krismer, B.; Peschel, A. Does Staphylococcus aureus nasal colonization involve biofilm formation? Future Microbiol. 2011, 6, 489–493. [Google Scholar] [CrossRef]
- Sugimoto, S.; Iwamoto, T.; Takada, K.; Okuda, K.; Tajima, A.; Iwase, T.; Mizunoe, Y. Staphylococcus epidermidis Esp degrades specific proteins associated with Staphylococcus aureus biofilm formation and host-pathogen interaction. J. Bacteriol. 2013, 195, 1645–1655. [Google Scholar] [CrossRef]
- Dubin, G.; Chmiel, D.; Mak, P.; Rakwalska, M.; Rzychon, M.; Dubin, A. Molecular cloning and biochemical characterisation of proteases from Staphylococcus epidermidis. Biol. Chem. 2001, 382, 1575–1582. [Google Scholar] [CrossRef]
- Glatthardt, T.; Campos, J.C.M.; Chamon, R.C.; de Sa Coimbra, T.F.; Rocha, G.A.; de Melo, M.A.F.; Parente, T.E.; Lobo, L.A.; Antunes, L.C.M.; Dos Santos, K.R.N.; et al. Small Molecules Produced by Commensal Staphylococcus epidermidis Disrupt Formation of Biofilms by Staphylococcus aureus. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef] [PubMed]
- Bode, L.G.; Kluytmans, J.A.; Wertheim, H.F.; Bogaers, D.; Vandenbroucke-Grauls, C.M.; Roosendaal, R.; Troelstra, A.; Box, A.T.; Voss, A.; van der Tweel, I.; et al. Preventing surgical-site infections in nasal carriers of Staphylococcus aureus. N. Engl. J. Med. 2010, 362, 9–17. [Google Scholar] [CrossRef]
- Wollenberg, M.S.; Claesen, J.; Escapa, I.F.; Aldridge, K.L.; Fischbach, M.A.; Lemon, K.P. Propionibacterium-produced coproporphyrin III induces Staphylococcus aureus aggregation and biofilm formation. mBio 2014, 5, e01286-14. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Loffler, B.; Proctor, R.A. Persistence of Staphylococcus aureus: Multiple Metabolic Pathways Impact the Expression of Virulence Factors in Small-Colony Variants (SCVs). Front. Microbiol. 2020, 11, 1028. [Google Scholar] [CrossRef]
- Proctor, R.A.; von Eiff, C.; Kahl, B.C.; Becker, K.; McNamara, P.; Herrmann, M.; Peters, G. Small colony variants: A pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 2006, 4, 295–305. [Google Scholar] [CrossRef]
- Onyango, L.A.; Hugh Dunstan, R.; Roberts, T.K.; Macdonald, M.M.; Gottfries, J. Phenotypic variants of staphylococci and their underlying population distributions following exposure to stress. PLoS ONE 2013, 8, e77614. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ray, P.; Das, A.; Sharma, M. Role of persisters and small-colony variants in antibiotic resistance of planktonic and biofilm-associated Staphylococcus aureus: An in vitro study. J. Med. Microbiol. 2009, 58, 1067–1073. [Google Scholar] [CrossRef]
- Cervantes-Garcia, E.; Garcia-Gonzalez, R.; Reyes-Torres, A.; Resendiz-Albor, A.A.; Salazar-Schettino, P.M. Staphylococcus aureus small colony variants in diabetic foot infections. Diabet. Foot Ankle 2015, 6, 26431. [Google Scholar] [CrossRef] [PubMed]
- Bui, L.M.G.; Kidd, S.P. A full genomic characterization of the development of a stable Small Colony Variant cell-type by a clinical Staphylococcus aureus strain. Infect. Genet. Evol. 2015, 36, 345–355. [Google Scholar] [CrossRef]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef]
- Guerillot, R.; Kostoulias, X.; Donovan, L.; Li, L.; Carter, G.P.; Hachani, A.; Vandelannoote, K.; Giulieri, S.; Monk, I.R.; Kunimoto, M.; et al. Unstable chromosome rearrangements in Staphylococcus aureus cause phenotype switching associated with persistent infections. Proc. Natl. Acad. Sci. USA 2019, 116, 20135–20140. [Google Scholar] [CrossRef]
- Proctor, R.A.; Peters, G. Small colony variants in staphylococcal infections: Diagnostic and therapeutic implications. Clin. Infect. Dis. 1998, 27, 419–422. [Google Scholar] [CrossRef] [PubMed]
- von Eiff, C.; McNamara, P.; Becker, K.; Bates, D.; Lei, X.H.; Ziman, M.; Bochner, B.R.; Peters, G.; Proctor, R.A. Phenotype microarray profiling of Staphylococcus aureus menD and hemB mutants with the small-colony-variant phenotype. J. Bacteriol. 2006, 188, 687–693. [Google Scholar] [CrossRef]
- Wong Fok Lung, T.; Monk, I.R.; Acker, K.P.; Mu, A.; Wang, N.; Riquelme, S.A.; Pires, S.; Noguera, L.P.; Dach, F.; Gabryszewski, S.J.; et al. Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat. Microbiol. 2020, 5, 141–153. [Google Scholar] [CrossRef]
- Clements, M.O.; Watson, S.P.; Poole, R.K.; Foster, S.J. CtaA of Staphylococcus aureus is required for starvation survival, recovery, and cytochrome biosynthesis. J. Bacteriol. 1999, 181, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Balwit, J.M.; van Langevelde, P.; Vann, J.M.; Proctor, R.A. Gentamicin-resistant menadione and hemin auxotrophic Staphylococcus aureus persist within cultured endothelial cells. J. Infect. Dis. 1994, 170, 1033–1037. [Google Scholar] [CrossRef]
- von Eiff, C.; Heilmann, C.; Proctor, R.A.; Woltz, C.; Peters, G.; Gotz, F. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J. Bacteriol. 1997, 179, 4706–4712. [Google Scholar] [CrossRef] [PubMed]
- Bogut, A.; Magrys, A. The road to success of coagulase-negative staphylococci: Clinical significance of small colony variants and their pathogenic role in persistent infections. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 2249–2270. [Google Scholar] [CrossRef]
- Vaudaux, P.; Francois, P.; Bisognano, C.; Kelley, W.L.; Lew, D.P.; Schrenzel, J.; Proctor, R.A.; McNamara, P.J.; Peters, G.; Von Eiff, C. Increased expression of clumping factor and fibronectin-binding proteins by hemB mutants of Staphylococcus aureus expressing small colony variant phenotypes. Infect. Immun. 2002, 70, 5428–5437. [Google Scholar] [CrossRef]
- Evans, M.D.; McDowell, S.A. Pleiotropic Effects of Statins: New Therapeutic Approaches to Chronic, Recurrent Infection by Staphylococcus aureus. Pharmaceutics 2021, 13, 2047. [Google Scholar] [CrossRef] [PubMed]
- Kahl, B.C. Small colony variants (SCVs) of Staphylococcus aureus—A bacterial survival strategy. Infect. Genet. Evol. 2014, 21, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Kahl, B.C.; Belling, G.; Reichelt, R.; Herrmann, M.; Proctor, R.A.; Peters, G. Thymidine-dependent small-colony variants of Staphylococcus aureus exhibit gross morphological and ultrastructural changes consistent with impaired cell separation. J. Clin. Microbiol. 2003, 41, 410–413. [Google Scholar] [CrossRef]
- Kahl, B.C.; Belling, G.; Becker, P.; Chatterjee, I.; Wardecki, K.; Hilgert, K.; Cheung, A.L.; Peters, G.; Herrmann, M. Thymidine-dependent Staphylococcus aureus small-colony variants are associated with extensive alterations in regulator and virulence gene expression profiles. Infect. Immun. 2005, 73, 4119–4126. [Google Scholar] [CrossRef]
- Fu, Z.; Tamber, S.; Memmi, G.; Donegan, N.P.; Cheung, A.L. Overexpression of MazFsa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J. Bacteriol. 2009, 191, 2051–2059. [Google Scholar] [CrossRef]
- Donegan, N.P.; Cheung, A.L. Regulation of the mazEF toxin-antitoxin module in Staphylococcus aureus and its impact on sigB expression. J. Bacteriol. 2009, 191, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Donegan, N.P.; Thompson, E.T.; Fu, Z.; Cheung, A.L. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 2010, 192, 1416–1422. [Google Scholar] [CrossRef]
- Brinsmade, S.R. CodY, a master integrator of metabolism and virulence in Gram-positive bacteria. Curr. Genet. 2017, 63, 417–425. [Google Scholar] [CrossRef]
- Siegmund, A.; Afzal, M.A.; Tetzlaff, F.; Keinhorster, D.; Gratani, F.; Paprotka, K.; Westermann, M.; Nietzsche, S.; Wolz, C.; Fraunholz, M.; et al. Intracellular persistence of Staphylococcus aureus in endothelial cells is promoted by the absence of phenol-soluble modulins. Virulence 2021, 12, 1186–1198. [Google Scholar] [CrossRef]
- Crosby, H.A.; Tiwari, N.; Kwiecinski, J.M.; Xu, Z.; Dykstra, A.; Jenul, C.; Fuentes, E.J.; Horswill, A.R. The Staphylococcus aureus ArlRS two-component system regulates virulence factor expression through MgrA. Mol. Microbiol. 2020, 113, 103–122. [Google Scholar] [CrossRef]
- Lee, J.; Zilm, P.S.; Kidd, S.P. Novel Research Models for Staphylococcus aureus Small Colony Variants (SCV) Development: Co-pathogenesis and Growth Rate. Front. Microbiol. 2020, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Mashruwala, A.A.; Guchte, A.V.; Boyd, J.M. Impaired respiration elicits SrrAB-dependent programmed cell lysis and biofilm formation in Staphylococcus aureus. eLife 2017, 6. [Google Scholar] [CrossRef]
- Mitchell, G.; Fugere, A.; Pepin Gaudreau, K.; Brouillette, E.; Frost, E.H.; Cantin, A.M.; Malouin, F. SigB is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS ONE 2013, 8, e65018. [Google Scholar] [CrossRef] [PubMed]
- Jenul, C.; Horswill, A.R. Regulation of Staphylococcus aureus Virulence. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Loffler, B.; Tuchscherr, L. Staphylococcus aureus Toxins: Promoter or Handicap during Infection? Toxins 2021, 13, 287. [Google Scholar] [CrossRef]
- Kriegeskorte, A.; Block, D.; Drescher, M.; Windmuller, N.; Mellmann, A.; Baum, C.; Neumann, C.; Lore, N.I.; Bragonzi, A.; Liebau, E.; et al. Inactivation of thyA in Staphylococcus aureus attenuates virulence and has a strong impact on metabolism and virulence gene expression. mBio 2014, 5, e01447-14. [Google Scholar] [CrossRef]
- Gao, W.; Chua, K.; Davies, J.K.; Newton, H.J.; Seemann, T.; Harrison, P.F.; Holmes, N.E.; Rhee, H.W.; Hong, J.I.; Hartland, E.L.; et al. Two novel point mutations in clinical Staphylococcus aureus reduce linezolid susceptibility and switch on the stringent response to promote persistent infection. PLoS Pathog. 2010, 6, e1000944. [Google Scholar] [CrossRef]
- Geiger, T.; Wolz, C. Intersection of the stringent response and the CodY regulon in low GC Gram-positive bacteria. Int. J. Med. Microbiol. 2014, 304, 150–155. [Google Scholar] [CrossRef]
- Hoffman, L.R.; Deziel, E.; D’Argenio, D.A.; Lepine, F.; Emerson, J.; McNamara, S.; Gibson, R.L.; Ramsey, B.W.; Miller, S.I. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2006, 103, 19890–19895. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Seguin, D.L.; Asselin, A.E.; Deziel, E.; Cantin, A.M.; Frost, E.H.; Michaud, S.; Malouin, F. Staphylococcus aureus sigma B-dependent emergence of small-colony variants and biofilm production following exposure to Pseudomonas aeruginosa 4-hydroxy-2-heptylquinoline-N-oxide. BMC Microbiol. 2010, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Biswas, L.; Biswas, R.; Schlag, M.; Bertram, R.; Gotz, F. Small-colony variant selection as a survival strategy for Staphylococcus aureus in the presence of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2009, 75, 6910–6912. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Kikuchi, K.; Nakamura, T.; Nakama, H.; Agematsu, K.; Kawakami, Y.; Maruchi, N.; Totsuka, K. H2O2 produced by viridans group streptococci may contribute to inhibition of methicillin-resistant Staphylococcus aureus colonization of oral cavities in newborns. Clin. Infect. Dis. 2001, 32, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Regev-Yochay, G.; Trzcinski, K.; Thompson, C.M.; Malley, R.; Lipsitch, M. Interference between Streptococcus pneumoniae and Staphylococcus aureus: In vitro hydrogen peroxide-mediated killing by Streptococcus pneumoniae. J. Bacteriol. 2006, 188, 4996–5001. [Google Scholar] [CrossRef]
- Wu, X.; Gordon, O.; Jiang, W.; Antezana, B.S.; Angulo-Zamudio, U.A.; Del Rio, C.; Moller, A.; Brissac, T.; Tierney, A.R.P.; Warncke, K.; et al. Interaction between Streptococcus pneumoniae and Staphylococcus aureus Generates .OH Radicals That Rapidly Kill Staphylococcus aureus Strains. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef]
- Painter, K.L.; Strange, E.; Parkhill, J.; Bamford, K.B.; Armstrong-James, D.; Edwards, A.M. Staphylococcus aureus adapts to oxidative stress by producing H2O2-resistant small-colony variants via the SOS response. Infect. Immun. 2015, 83, 1830–1844. [Google Scholar] [CrossRef]
- Margolis, E. Hydrogen peroxide-mediated interference competition by Streptococcus pneumoniae has no significant effect on Staphylococcus aureus nasal colonization of neonatal rats. J. Bacteriol. 2009, 191, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Hardy, B.L.; Dickey, S.W.; Plaut, R.D.; Riggins, D.P.; Stibitz, S.; Otto, M.; Merrell, D.S. Corynebacterium pseudodiphtheriticum Exploits Staphylococcus aureus Virulence Components in a Novel Polymicrobial Defense Strategy. mBio 2019, 10, e02491-18. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, J.M.; Bartels, D.J.; Volper, E.M.; Greenberg, E.P. Quorum sensing in Staphylococcus aureus biofilms. J. Bacteriol. 2004, 186, 1838–1850. [Google Scholar] [CrossRef]
- Jenkins, A.; Diep, B.A.; Mai, T.T.; Vo, N.H.; Warrener, P.; Suzich, J.; Stover, C.K.; Sellman, B.R. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. mBio 2015, 6, e02272-14. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef] [PubMed]
- Geisinger, E.; Isberg, R.R. Interplay Between Antibiotic Resistance and Virulence During Disease Promoted by Multidrug-Resistant Bacteria. J. Infect. Dis. 2017, 215, S9–S17. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Davis, M.H.; Peterson, J.; Solis-Lemus, C.; Satola, S.W.; Read, T.D. Effect of genetic background on the evolution of Vancomycin-Intermediate Staphylococcus aureus (VISA). PeerJ 2021, 9, e11764. [Google Scholar] [CrossRef]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281. [Google Scholar]
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Investig. 2003, 111, 1265–1273. [Google Scholar] [CrossRef]
- Reygaert, W.C. Antimicrobial Resistance Mechanisms of Staphylococcus aureus; Formatex Research Center: Norristown, PA, USA, 2013. [Google Scholar]
- Petit, R.A., 3rd; Read, T.D. Staphylococcus aureus viewed from the perspective of 40,000+ genomes. PeerJ 2018, 6, e5261. [Google Scholar] [CrossRef]
- Mirzaie, A.; Peirovi, N.; Akbarzadeh, I.; Moghtaderi, M.; Heidari, F.; Yeganeh, F.E.; Noorbazargan, H.; Mirzazadeh, S.; Bakhtiari, R. Preparation and optimization of ciprofloxacin encapsulated niosomes: A new approach for enhanced antibacterial activity, biofilm inhibition and reduced antibiotic resistance in ciprofloxacin-resistant methicillin-resistance Staphylococcus aureus. Bioorg Chem. 2020, 103, 104231. [Google Scholar] [CrossRef] [PubMed]
- Westblade, L.F.; Errington, J.; Dorr, T. Antibiotic tolerance. PLoS Pathog. 2020, 16, e1008892. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.S.; Berryhill, B.A.; Gil-Gil, T.; Manuel, J.A.; Smith, A.P.; Baquero, F.; Levin, B.R. The Tradeoffs Between Persistence and Mutation Rates at Sub-Inhibitory Antibiotic Concentrations in Staphylococcus aureus. bioRxiv 2024. [Google Scholar] [CrossRef]
- Elgrail, M.M.; Chen, E.; Shaffer, M.G.; Srinivasa, V.; Griffith, M.P.; Mustapha, M.M.; Shields, R.K.; Van Tyne, D.; Culyba, M.J. Convergent Evolution of Antibiotic Tolerance in Patients with Persistent Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect. Immun. 2022, 90, e0000122. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, K.; Zhang, H.; Jia, Y.; Wang, Z. Combating Antibiotic Tolerance Through Activating Bacterial Metabolism. Front. Microbiol. 2020, 11, 577564. [Google Scholar] [CrossRef] [PubMed]
- Salzer, A.; Ingrassia, S.; Sauer, L.; Rapp, J.; Dobritz, R.; Müller, J.; Link, H.; Wolz, C. (p)ppGpp-mediated GTP homeostasis ensures the survival and antibiotic tolerance of Staphylococcus aureus during starvation by preserving the proton motive force. bioRxiv 2024. [Google Scholar] [CrossRef]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef]
- Coelho, C.; de Lencastre, H.; Aires-de-Sousa, M. Frequent occurrence of trimethoprim-sulfamethoxazole hetero-resistant Staphylococcus aureus isolates in different African countries. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1243–1252. [Google Scholar] [CrossRef]
- Manjunath, A.; Thumu, S.C.R.; Kumar, S.; Halami, P.M. Bacterial heteroresistance: An evolving novel way to combat antibiotics. Biologia 2021, 76, 3029–3041. [Google Scholar] [CrossRef]
- Abavisani, M.; Kodori, M.; Akrami, F.; Radfar, A.; Hashemi, A. Relationships between Efflux Pumps and Emergence of Heteroresistance: A Comprehensive Study on the Current Findings. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 3916980. [Google Scholar] [CrossRef]
- Band, V.I.; Weiss, D.S. Heteroresistance: A cause of unexplained antibiotic treatment failure? PLoS Pathog. 2019, 15, e1007726. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, L.; Fauvart, M.; Michiels, J. Bacterial Heterogeneity and Antibiotic Survival: Understanding and Combatting Persistence and Heteroresistance. Mol. Cell 2019, 76, 255–267. [Google Scholar] [CrossRef]
- El-Halfawy, O.M.; Valvano, M.A. Antimicrobial heteroresistance: An emerging field in need of clarity. Clin. Microbiol. Rev. 2015, 28, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Nicoloff, H.; Hjort, K. Mechanisms and clinical relevance of bacterial heteroresistance. Nat. Rev. Microbiol. 2019, 17, 479–496. [Google Scholar] [CrossRef]
- Al-Shebiny, A.; Shawky, R.; Emara, M. Heteroresistance: A Gray Side of Antimicrobial Susceptibility Testing. J. Adv. Pharm. Res. 2023, 7, 101–110. [Google Scholar] [CrossRef]
- Hjort, K.; Nicoloff, H.; Andersson, D.I. Unstable tandem gene amplification generates heteroresistance (variation in resistance within a population) to colistin in Salmonella enterica. Mol. Microbiol. 2016, 102, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Nicoloff, H.; Hjort, K.; Levin, B.R.; Andersson, D.I. The high prevalence of antibiotic heteroresistance in pathogenic bacteria is mainly caused by gene amplification. Nat. Microbiol. 2019, 4, 504–514. [Google Scholar] [CrossRef]
- Sandegren, L.; Andersson, D.I. Bacterial gene amplification: Implications for the evolution of antibiotic resistance. Nat. Rev. Microbiol. 2009, 7, 578–588. [Google Scholar] [CrossRef]
- Pereira, C.; Larsson, J.; Hjort, K.; Elf, J.; Andersson, D.I. The highly dynamic nature of bacterial heteroresistance impairs its clinical detection. Commun. Biol. 2021, 4, 521. [Google Scholar] [CrossRef]
- Heidarian, S.; Guliaev, A.; Nicoloff, H.; Hjort, K.; Andersson, D.I. High prevalence of heteroresistance in Staphylococcus aureus is caused by a multitude of mutations in core genes. PLoS Biol. 2024, 22, e3002457. [Google Scholar] [CrossRef]
- Weiner-Lastinger, L.M.; Abner, S.; Edwards, J.R.; Kallen, A.J.; Karlsson, M.; Magill, S.S.; Pollock, D.; See, I.; Soe, M.M.; Walters, M.S.; et al. Antimicrobial-resistant pathogens associated with adult healthcare-associated infections: Summary of data reported to the National Healthcare Safety Network, 2015–2017. Infect. Control Hosp. Epidemiol. 2020, 41, 1–18. [Google Scholar] [CrossRef]
- Davis, K.A.; Stewart, J.J.; Crouch, H.K.; Florez, C.E.; Hospenthal, D.R. Methicillin-resistant Staphylococcus aureus (MRSA) nares colonization at hospital admission and its effect on subsequent MRSA infection. Clin. Infect. Dis. 2004, 39, 776–782. [Google Scholar] [CrossRef]
- Kalmeijer, M.D.; van Nieuwland-Bollen, E.; Bogaers-Hofman, D.; de Baere, G.A. Nasal carriage of Staphylococcus aureus is a major risk factor for surgical-site infections in orthopedic surgery. Infect. Control Hosp. Epidemiol. 2000, 21, 319–323. [Google Scholar] [CrossRef]
- Septimus, E.J.; Schweizer, M.L. Decolonization in Prevention of Health Care-Associated Infections. Clin. Microbiol. Rev. 2016, 29, 201–222. [Google Scholar] [CrossRef]
- Gagnaire, J.; Botelho-Nevers, E.; Martin-Simoes, P.; Morel, J.; Zeni, F.; Maillard, N.; Mariat, C.; Haddar, C.H.; Carricajo, A.; Fonsale, N.; et al. Interplay of nasal and rectal carriage of Staphylococcus aureus in intensive care unit patients. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Haleem, A.; Schultz, J.S.; Heilmann, K.P.; Dohrn, C.L.; Diekema, D.J.; Gardner, S.E. Concordance of nasal and diabetic foot ulcer staphylococcal colonization. Diagn. Microbiol. Infect. Dis. 2014, 79, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Zukowska, A.; Zukowski, M. Surgical Site Infection in Cardiac Surgery. J. Clin. Med. 2022, 11, 6991. [Google Scholar] [CrossRef]
- Lavigne, J.P.; Hosny, M.; Dunyach-Remy, C.; Boutet-Dubois, A.; Schuldiner, S.; Cellier, N.; Yahiaoui-Martinez, A.; Molle, V.; La Scola, B.; Marchandin, H.; et al. Long-Term Intrahost Evolution of Staphylococcus aureus Among Diabetic Patients With Foot Infections. Front. Microbiol. 2021, 12, 741406. [Google Scholar] [CrossRef]
- Dunyach-Remy, C.; Courtais-Coulon, C.; DeMattei, C.; Jourdan, N.; Schuldiner, S.; Sultan, A.; Carriere, C.; Alonso, S.; Sotto, A.; Lavigne, J.P. Link between nasal carriage of Staphylococcus aureus and infected diabetic foot ulcers. Diabetes Metab. 2017, 43, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Gjodsbol, K.; Skindersoe, M.E.; Skov, R.L.; Krogfelt, K.A. Cross-contamination: Comparison of Nasal and Chronic Leg Ulcer Staphylococcus aureus Strains Isolated from the Same Patient. Open Microbiol. J. 2013, 7, 6–8. [Google Scholar] [CrossRef]
- Hill, R.L. The bioavailability of mupirocin in nasal secretions in vitro. J. Clin. Pathol. 2002, 55, 233–235. [Google Scholar] [CrossRef]
- Stanaway, S.; Johnson, D.; Moulik, P.; Gill, G. Methicillin-resistant Staphylococcus aureus (MRSA) isolation from diabetic foot ulcers correlates with nasal MRSA carriage. Diabetes Res. Clin. Pract. 2007, 75, 47–50. [Google Scholar] [CrossRef]
- Besier, S.; Smaczny, C.; von Mallinckrodt, C.; Krahl, A.; Ackermann, H.; Brade, V.; Wichelhaus, T.A. Prevalence and clinical significance of Staphylococcus aureus small-colony variants in cystic fibrosis lung disease. J. Clin. Microbiol. 2007, 45, 168–172. [Google Scholar] [CrossRef]
- Neut, D.; van der Mei, H.C.; Bulstra, S.K.; Busscher, H.J. The role of small-colony variants in failure to diagnose and treat biofilm infections in orthopedics. Acta Orthop. 2007, 78, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Fugere, A.; Lalonde Seguin, D.; Mitchell, G.; Deziel, E.; Dekimpe, V.; Cantin, A.M.; Frost, E.; Malouin, F. Interspecific small molecule interactions between clinical isolates of Pseudomonas aeruginosa and Staphylococcus aureus from adult cystic fibrosis patients. PLoS ONE 2014, 9, e86705. [Google Scholar] [CrossRef]
- Kahl, B.; Herrmann, M.; Everding, A.S.; Koch, H.G.; Becker, K.; Harms, E.; Proctor, R.A.; Peters, G. Persistent infection with small colony variant strains of Staphylococcus aureus in patients with cystic fibrosis. J. Infect. Dis. 1998, 177, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Orazi, G.; O’Toole, G.A. Pseudomonas aeruginosa Alters Staphylococcus aureus Sensitivity to Vancomycin in a Biofilm Model of Cystic Fibrosis Infection. mBio 2017, 8, e00873-17. [Google Scholar] [CrossRef] [PubMed]
- Suwantarat, N.; Rubin, M.; Bryan, L.; Tekle, T.; Boyle, M.P.; Carroll, K.C.; Jennings, M.T. Frequency of small-colony variants and antimicrobial susceptibility of methicillin-resistant Staphylococcus aureus in cystic fibrosis patients. Diagn. Microbiol. Infect. Dis. 2018, 90, 296–299. [Google Scholar] [CrossRef]
- Masoud-Landgraf, L.; Zarfel, G.; Kaschnigg, T.; Friedl, S.; Feierl, G.; Wagner-Eibel, U.; Eber, E.; Grisold, A.J.; Kittinger, C. Analysis and Characterization of Staphylococcus aureus Small Colony Variants Isolated from Cystic Fibrosis Patients in Austria. Curr. Microbiol. 2016, 72, 606–611. [Google Scholar] [CrossRef]
- Keim, K.C.; George, I.K.; Reynolds, L.; Smith, A.C. The Clinical Significance of Staphylococcus aureus Small Colony Variants. Lab. Med. 2023, 54, 227–234. [Google Scholar] [CrossRef]
- Palavecino, E.L. Rapid Methods for Detection of MRSA in Clinical Specimens. Methods Mol. Biol. 2020, 2069, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Coombs, G.W.; Daley, D.A.; Mowlaboccus, S.; Pang, S.; Australian Group on Antimicrobial, R. Australian Group on Antimicrobial Resistance (AGAR) Australian Enterococcal Sepsis Outcome Programme (AESOP) Annual Report 2019. Commun. Dis. Intell. (2018) 2020, 44, 1–12. [Google Scholar] [CrossRef]
- Vaudaux, P.; Kelley, W.L.; Lew, D.P. Staphylococcus aureus small colony variants: Difficult to diagnose and difficult to treat. Clin. Infect. Dis. 2006, 43, 968–970. [Google Scholar] [CrossRef]
- von Eiff, C.; Peters, G.; Becker, K. The small colony variant (SCV) concept—The role of staphylococcal SCVs in persistent infections. Injury 2006, 37 (Suppl. S2), S26–S33. [Google Scholar] [CrossRef] [PubMed]
- Kipp, F.; Kahl, B.C.; Becker, K.; Baron, E.J.; Proctor, R.A.; Peters, G.; von Eiff, C. Evaluation of two chromogenic agar media for recovery and identification of Staphylococcus aureus small-colony variants. J. Clin. Microbiol. 2005, 43, 1956–1959. [Google Scholar] [CrossRef] [PubMed]
- Melter, O.; Radojevic, B. Small colony variants of Staphylococcus aureus—Review. Folia Microbiol. 2010, 55, 548–558. [Google Scholar] [CrossRef]
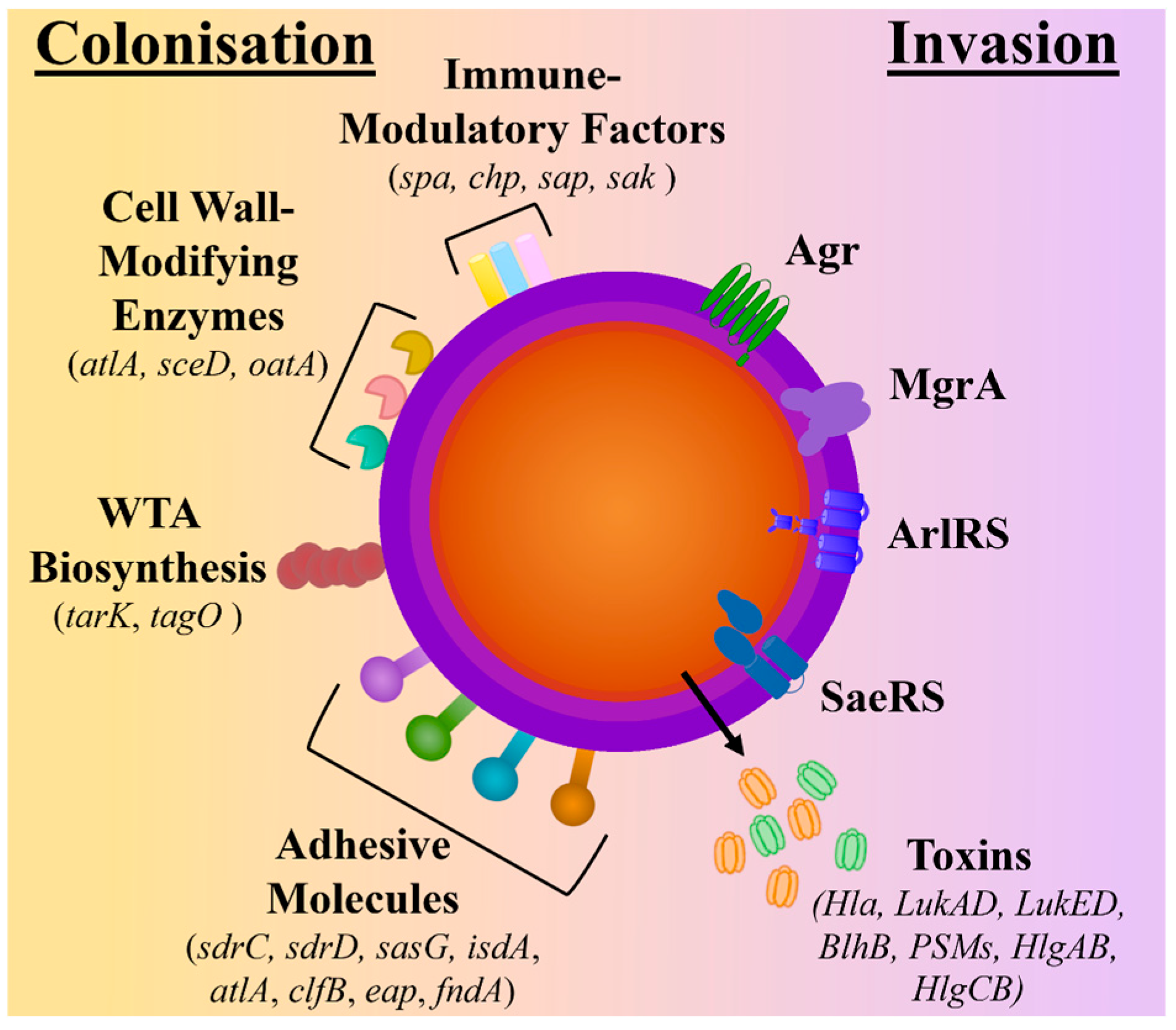
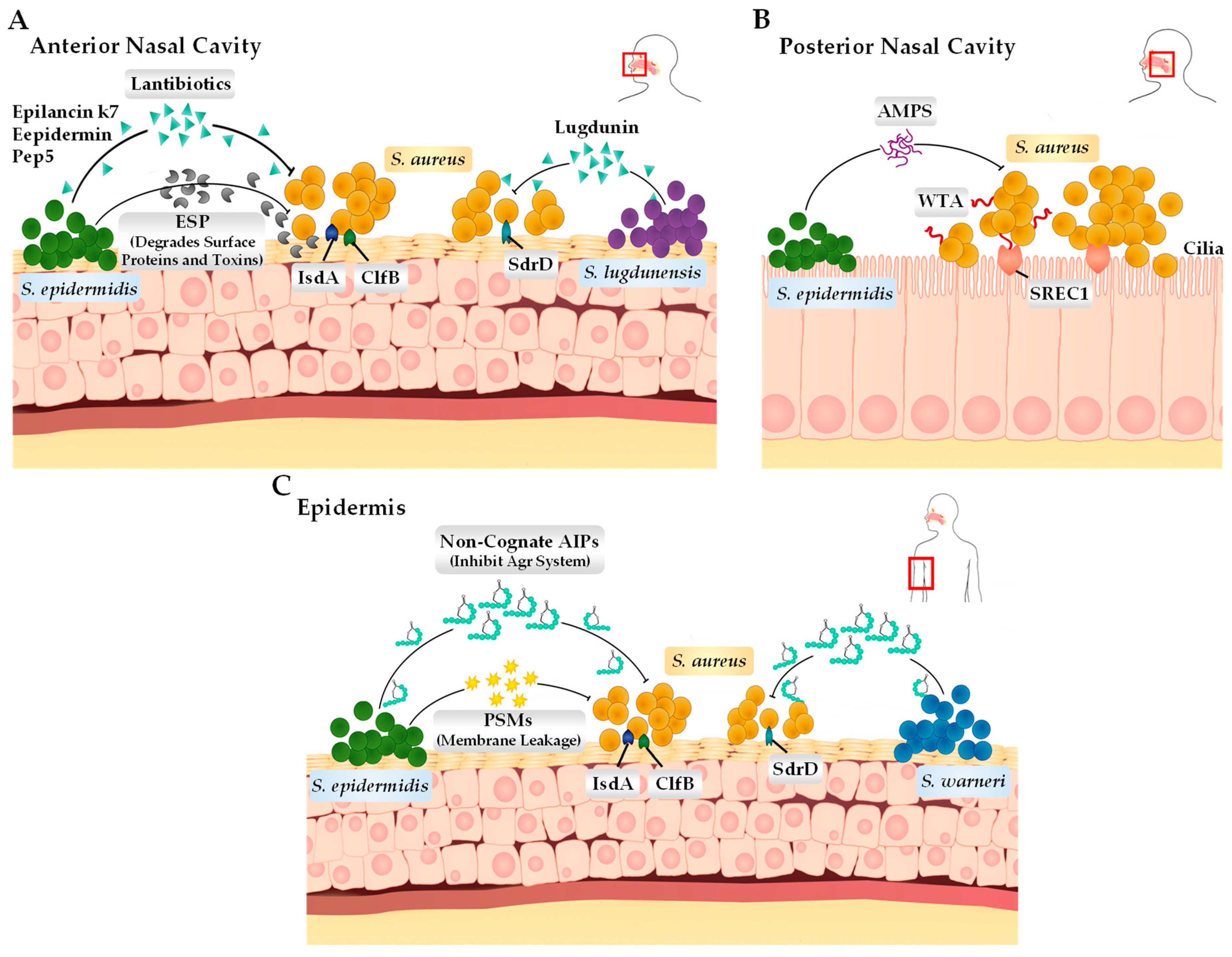

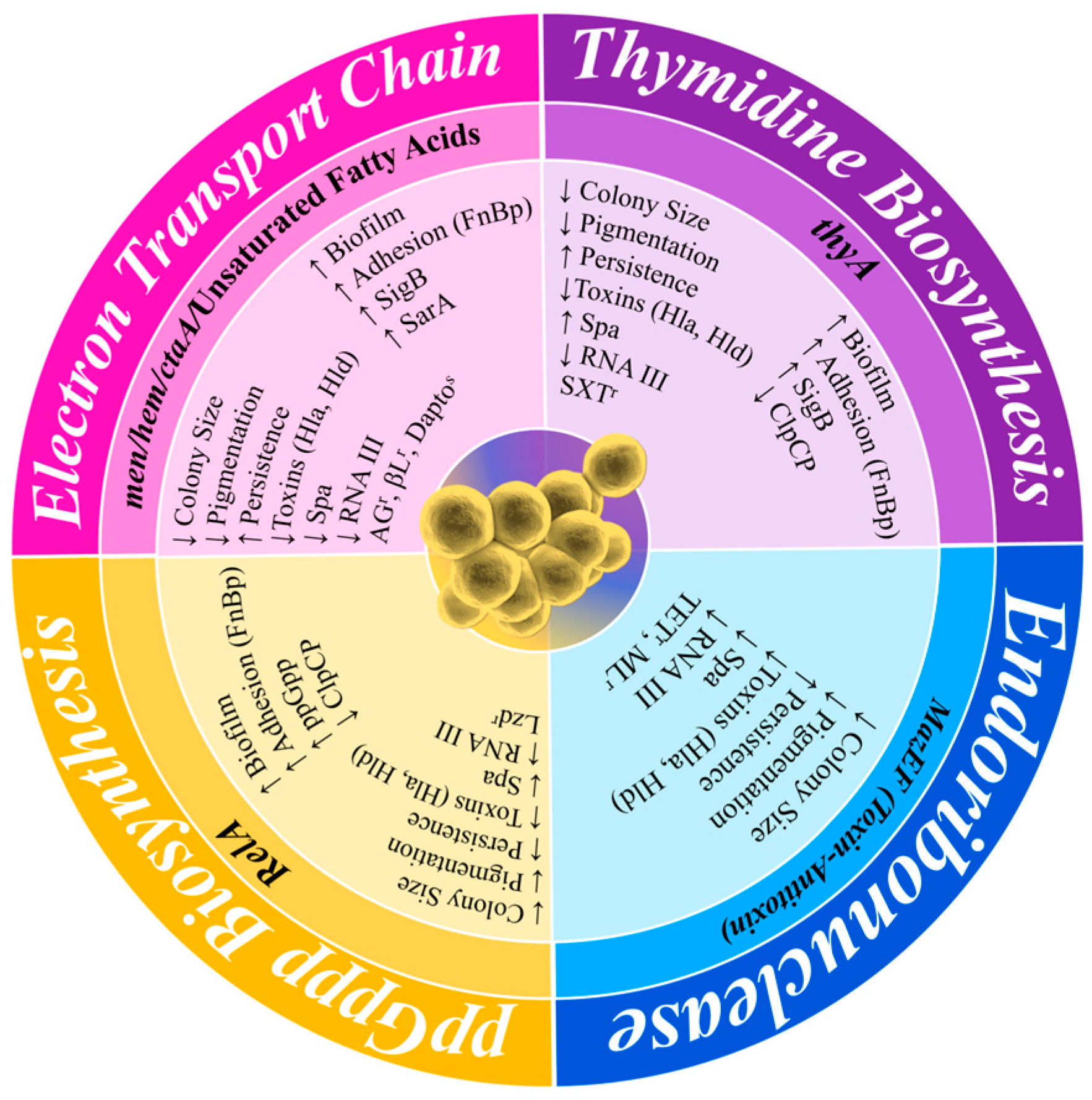
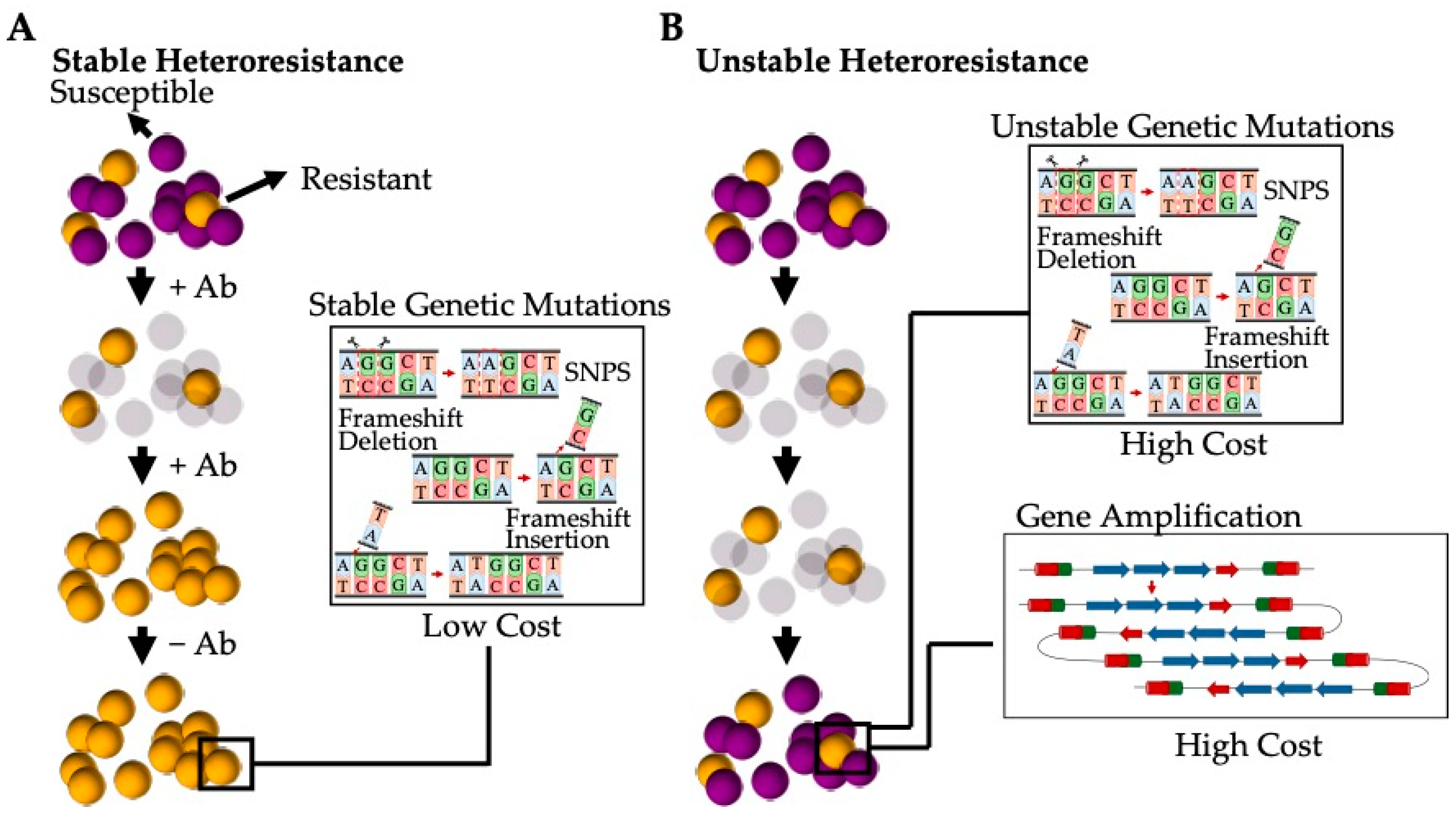
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).