
Purification of a Multidrug Resistance Transporter for Crystallization Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
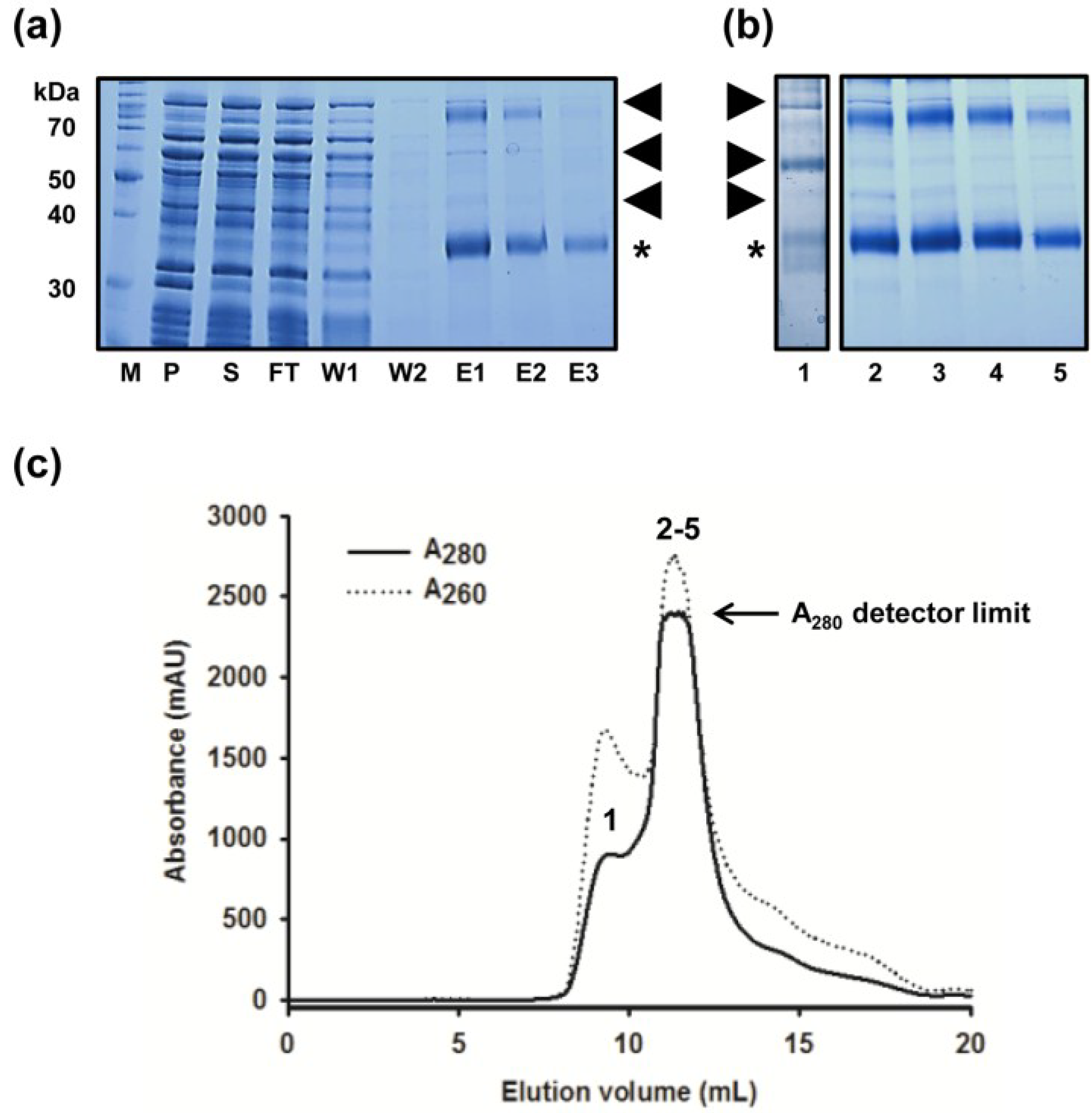
2.1. Improved Purification of MdtM and Identification of Key Contaminant


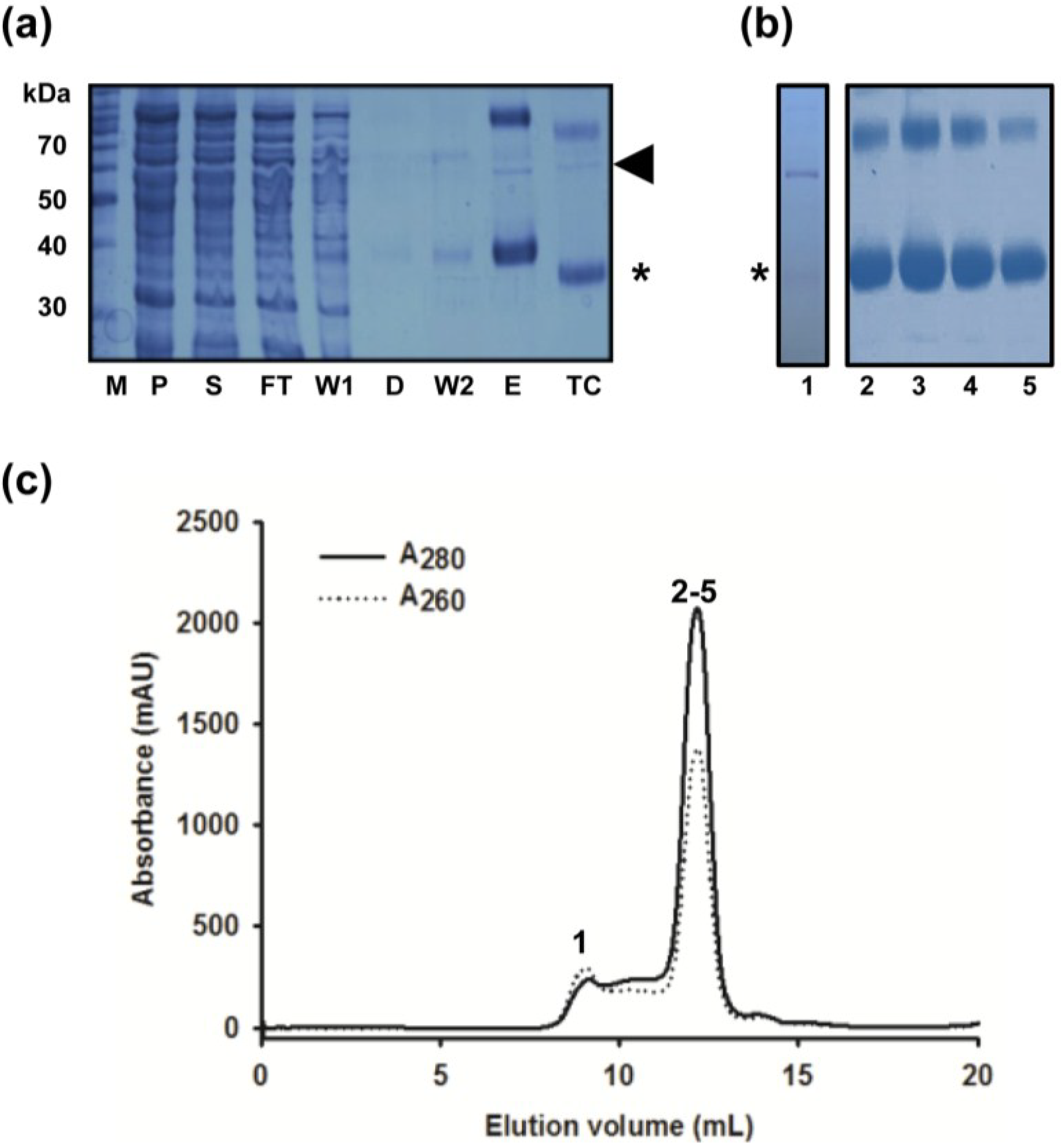
2.2. Removal of Chaperonin Contamination
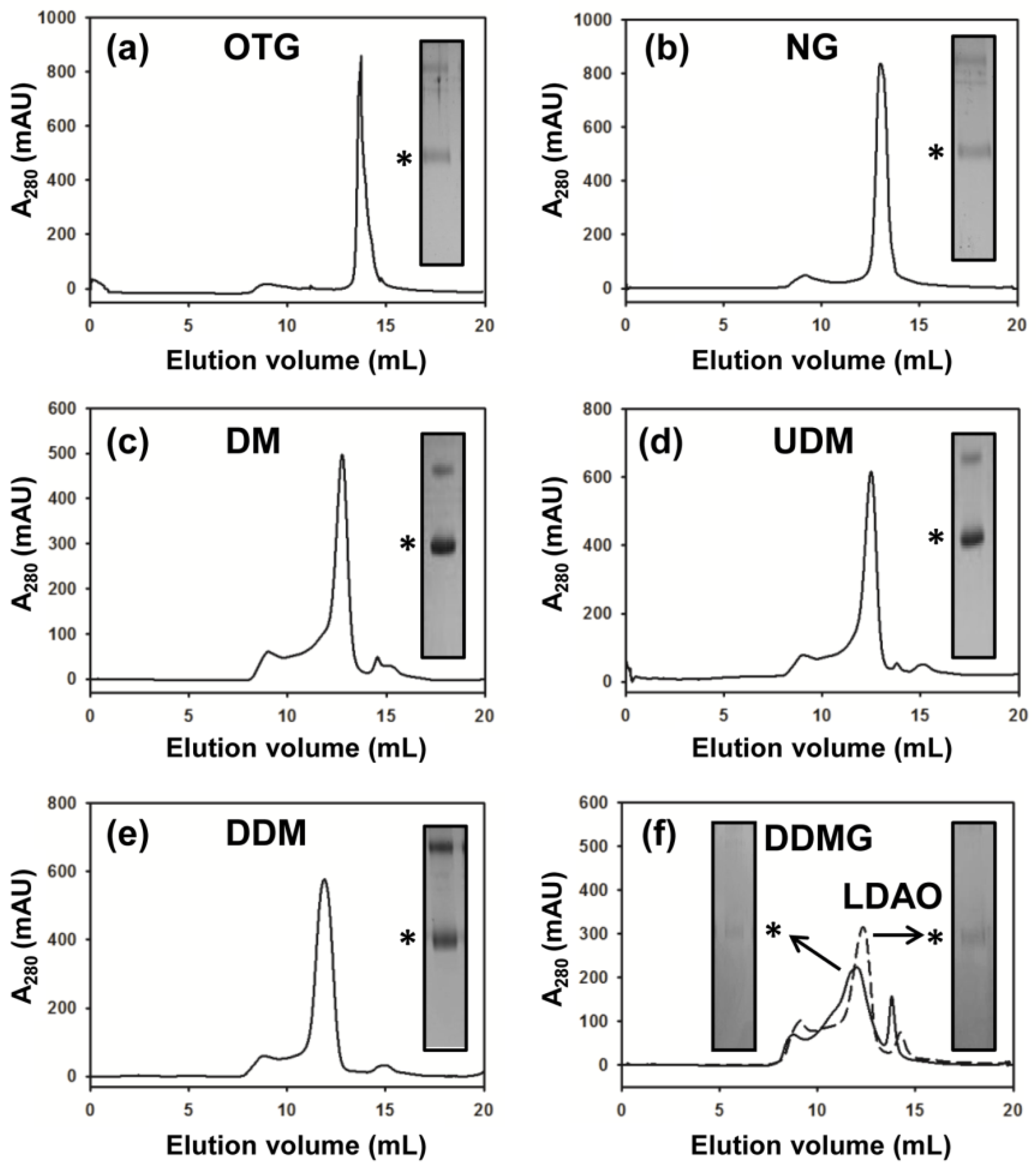
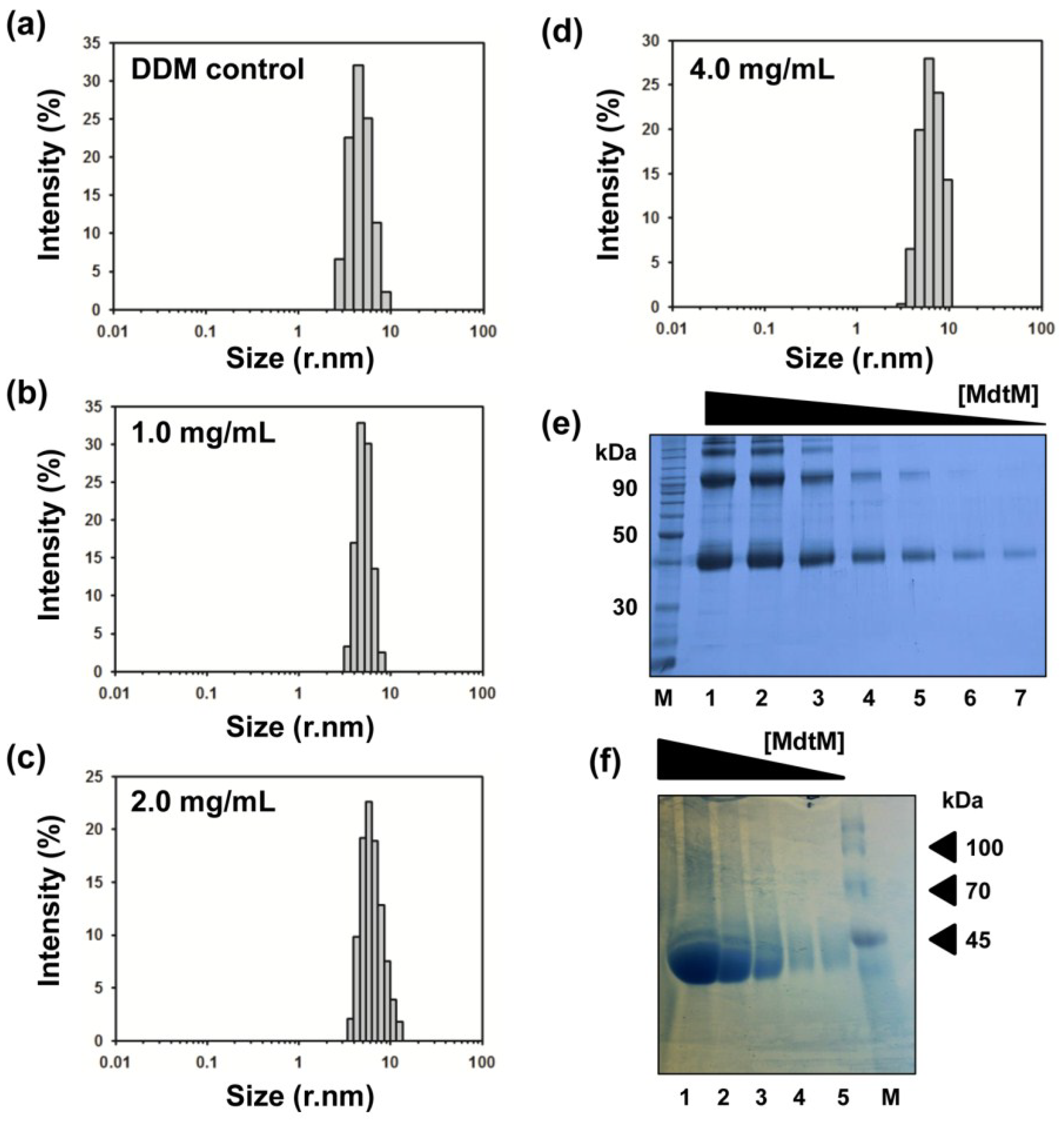
2.3. DM Is the Shortest Alkyl-Chain Detergent that Maintains MdtM in a Stable and Monodispersed State

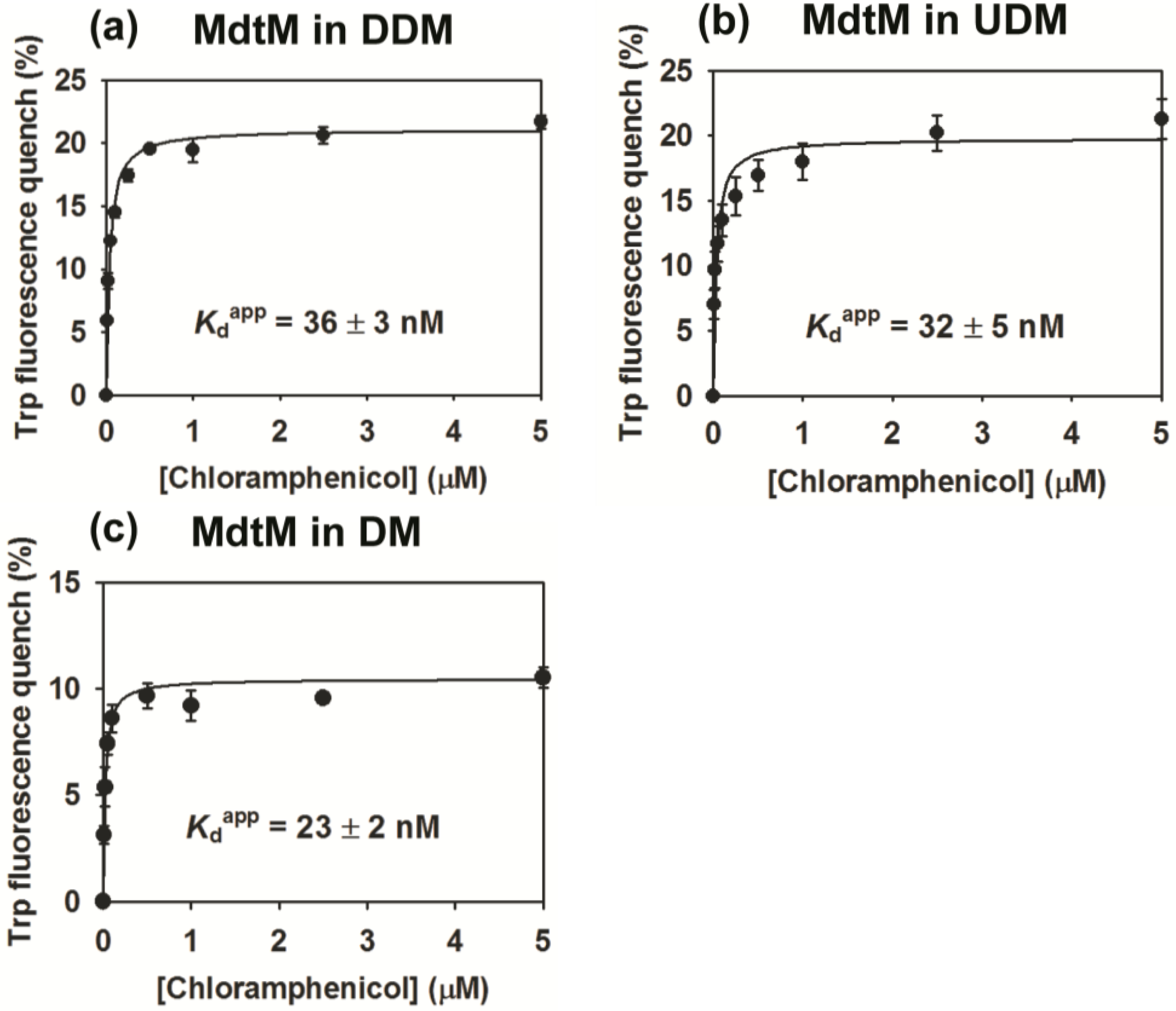
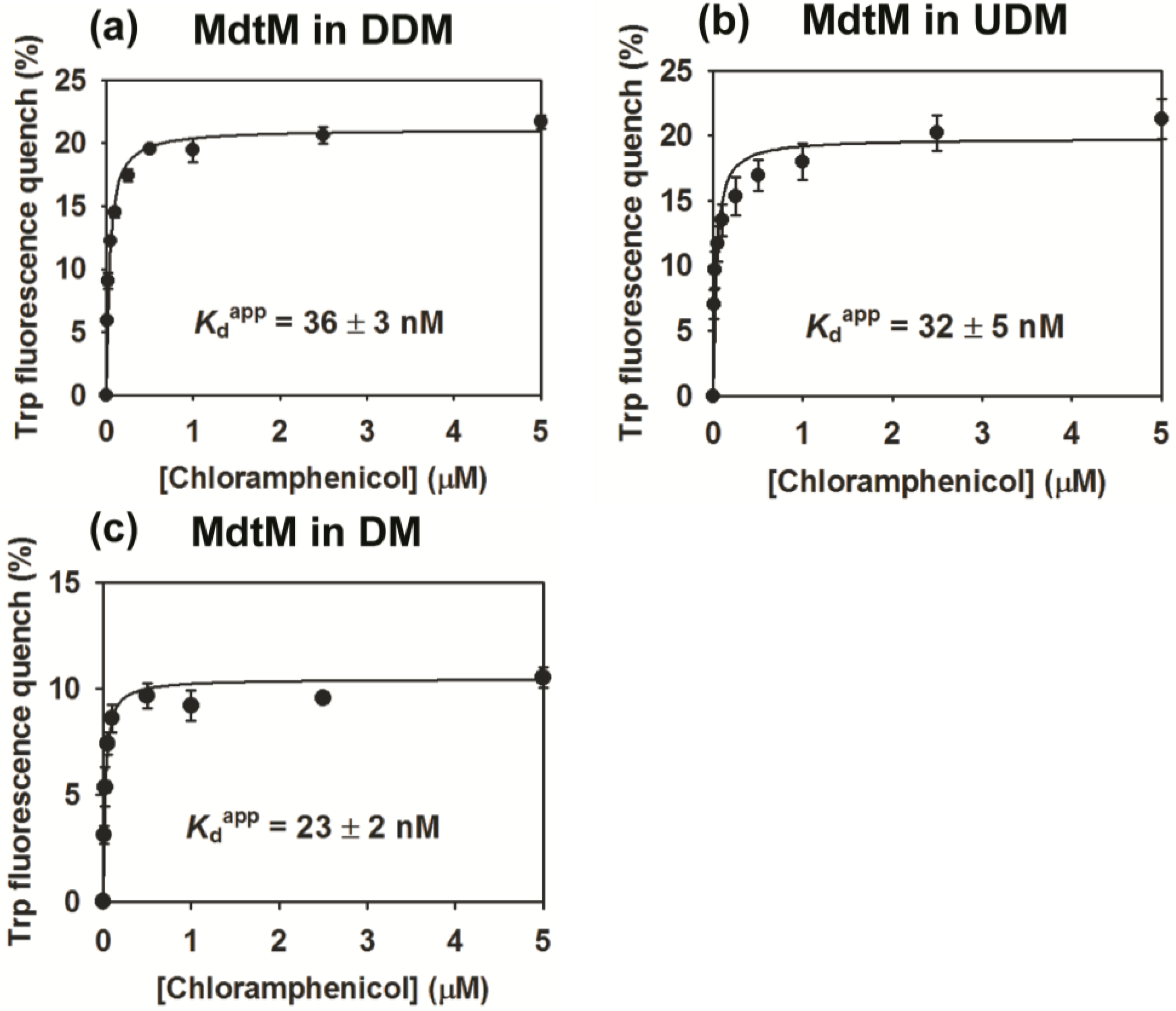
2.4. Binding Affinity of Purified MdtM
2.5. Determination of the Oligomeric State of MdtM

2.6. Thermostability of MdtM in DDM, UDM and DM

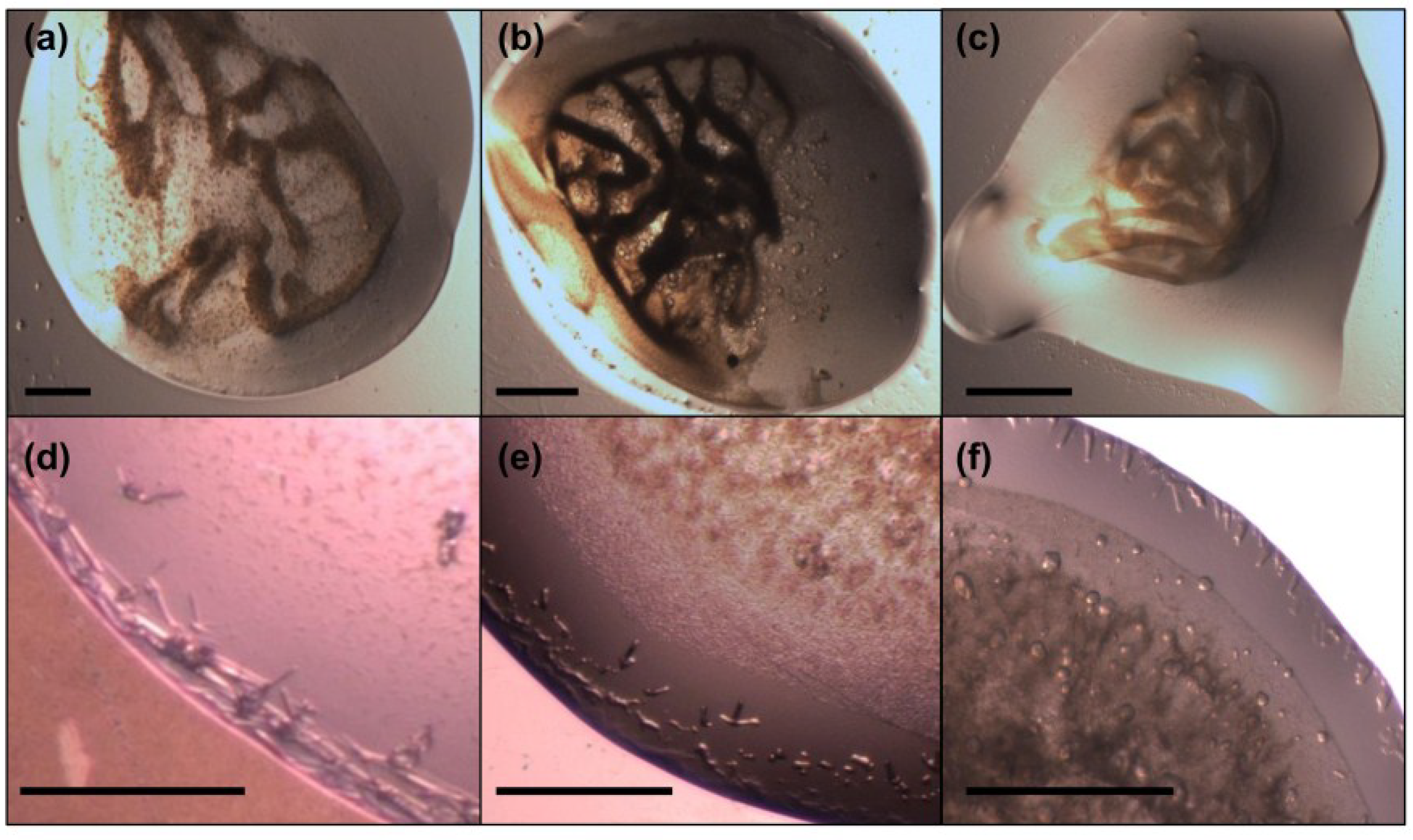
2.7. Crystallization of MdtM

3. Experimental Section
3.1. Materials
3.2. Cloning, Plasmids and Bacterial Strains
3.3. Overproduction of MdtM and Membrane Preparation
3.4. Immobilized Metal Affinity Chromatography
3.5. Size Exclusion Chromatography
3.6. Detergent Screening
3.7. Substrate Binding Assays
3.8. Thermostability Assays
3.9. Mass Spectrometry
3.10. Dynamic Light Scattering
3.11. Blue Native PAGE
3.12. Crystallization of MdtM
4. Conclusions
Acknowledgments
Author Contributions
Appendix

Conflicts of Interest
References
- Putman, M.; van Veen, H.W.; Konings, W.N. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 2000, 64, 672–693. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, C.; Michel, H. Crystallization of membrane proteins. Curr. Opin. Struct. Biol. 1997, 7, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.N.; Safferling, M.; Lemieux, M.J.; Griffith, H.; Chen, Y.; Li, X.D. Practical aspects of overexpressing bacterial secondary membrane transporters for structural studies. Biochim. Biophys. Acta 2003, 1610, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Shlykov, M.A.; Castillo, R.; Sun, E.I.; Saier, M.H., Jr. The major facilitator superfamily (MFS) revisited. FEBS J. 2012, 279, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lemieux, M.J.; Song, J.; Auer, M.; Wang, D.N. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003, 301, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Smirnova, I.; Kasho, V.; Verner, G.; Kaback, H.R.; Iwata, S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003, 301, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; He, X.; Szewczyk, P.; Nguyen, T.; Chang, G. Structure of the multidrug transporter EmrD from Escherichia coli. Science 2006, 312, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.; Sun, L.; Huang, Y.; Lu, F.; Liu, Y.; Gong, H.; Wang, J.; Yan, N. Structure of a fucose transporter in an outward-open conformation. Nature 2010, 467, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Newstead, S.; Drew, D.; Cameron, A.D.; Postis, V.L.; Xia, X.; Fowler, P.W.; Ingram, J.C.; Carpenter, E.P.; Sansom, M.S.; McPherson, M.J.; et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011, 30, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Ethayathulla, A.S.; Yousef, M.S.; Amin, A.; Leblanc, G.; Kaback, H.R.; Guan, L. Structure-based mechanism for Na+/melibiose symport by MelB. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, Y.; Wang, X.; Fan, J.; Heng, J.; Liu, X.; Feng, W.; Kang, X.; Huang, B.; Liu, J.; et al. Structure of the yajr transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. USA 2013, 110, 14664–14669. [Google Scholar] [CrossRef] [PubMed]
- Iancu, C.V.; Zamoon, J.; Woo, S.B.; Aleshin, A.; Choe, J.Y. Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc. Natl. Acad. Sci. USA 2013, 110, 17862–17867. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Wisedchaisri, G.; Gonen, T. Crystal structure of a nitrate/nitrite exchanger. Nature 2013, 497, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Quistgaard, E.M.; Low, C.; Moberg, P.; Tresaugues, L.; Nordlund, P. Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat. Struct. Mol. Biol. 2013, 20, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Huang, W.; Yan, C.; Gong, X.; Jiang, S.; Zhao, Y.; Wang, J.; Shi, Y. Structure and mechanism of a nitrate transporter. Cell Rep. 2013, 3, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.P.; Kumar, H.; Waight, A.B.; Risenmay, A.J.; Roe-Zurz, Z.; Chau, B.H.; Schlessinger, A.; Bonomi, M.; Harries, W.; Sali, A.; et al. Crystal structure of a eukaryotic phosphate transporter. Nature 2013, 496, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Newstead, S. Molecular basis of nitrate uptake by the plant nitrate transporter Nrt1.1. Nature 2014, 507, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr.; Beatty, J.T.; Goffeau, A.; Harley, K.T.; Heijne, W.H.; Huang, S.C.; Jack, D.L.; Jahn, P.S.; Lew, K.; Liu, J.; et al. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1999, 1, 257–279. [Google Scholar] [PubMed]
- Holdsworth, S.R.; Law, C.J. Functional and biochemical characterisation of the Escherichia coli major facilitator superfamily multidrug transporter MdtM. Biochimie 2012, 94, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, S.R.; Law, C.J. The major facilitator superfamily transporter MdtM contributes to the intrinsic resistance of Escherichia coli to quaternary ammonium compounds. J. Antimicrob. Chemother. 2013, 68, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, S.R.; Law, C.J. Multidrug resistance protein MdtM adds to the repertoire of antiporters involved in alkaline pH homeostasis in Escherichia coli. BMC Microbiol. 2013. [Google Scholar] [CrossRef]
- Paul, S.; Alegre, K.O.; Holdsworth, S.R.; Rice, M.; Brown, J.A.; McVeigh, P.; Kelly, S.M.; Law, C.J. A single-component multidrug transporter of the major facilitator superfamily is part of a network that protects Escherichia coli from bile salt stress. Mol. Microbiol. 2014, 92, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Bibi, E. MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J. Bacteriol. 1997, 179, 2274–2280. [Google Scholar] [PubMed]
- Edgar, R.; Bibi, E. A single membrane-embedded negative charge is critical for recognizing positively charged drugs by the Escherichia coli multidrug resistance protein MdfA. EMBO J. 1999, 18, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Lewinson, O.; Bibi, E. Evidence for simultaneous binding of dissimilar substrates by the Escherichia coli multidrug transporter MdfA. Biochemistry 2001, 40, 12612–12618. [Google Scholar] [CrossRef] [PubMed]
- Lewinson, O.; Adler, J.; Poelarends, G.J.; Mazurkiewicz, P.; Driessen, A.J.; Bibi, E. The Escherichia coli multidrug transporter MdfA catalyzes both electrogenic and electroneutral transport reactions. Proc. Natl. Acad. Sci. USA 2003, 100, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Lewinson, O.; Adler, J.; Sigal, N.; Bibi, E. Promiscuity in multidrug recognition and transport: The bacterial MFS Mdr transporters. Mol. Microbiol. 2006, 61, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Fluman, N.; Ryan, C.M.; Whitelegge, J.P.; Bibi, E. Dissection of mechanistic principles of a secondary multidrug efflux protein. Mol. Cell 2012, 47, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.; Bibi, E. Determinants of substrate recognition by the Escherichia coli multidrug transporter MdfA identified on both sides of the membrane. J. Biol. Chem. 2004, 279, 8957–8965. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.; Bibi, E. Promiscuity in the geometry of electrostatic interactions between the Escherichia coli multidrug resistance transporter MdfA and cationic substrates. J. Biol. Chem. 2005, 280, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.; Lewinson, O.; Bibi, E. Role of a conserved membrane-embedded acidic residue in the multidrug transporter MdfA. Biochemistry 2004, 43, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, S.R. Purification and Characterisation of the Escherichia coli Multidrug Efflux Protein MdtM. Ph.D. Thesis, Queen’s University Belfast, Belfast, Northern Ireland, 2013. [Google Scholar]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [PubMed]
- Auer, M.; Kim, M.J.; Lemieux, M.J.; Villa, A.; Song, J.; Li, X.D.; Wang, D.N. High-yield expression and functional analysis of Escherichia coli glycerol-3-phosphate transporter. Biochemistry 2001, 40, 6628–6635. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Ito, T. Absorption spectra of deoxyribose, ribosephosphate, ATP and DNA by direct transmission measurements in the vacuum-UV (150–190 nm) and far-UV (190–260 nm) regions using synchrotron radiation as a light source. Photochem. Photobiol. 1986, 44, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.E.; Andreotti, A.H. Bacterial expression and purification of interleukin-2 tyrosine kinase: Single step separation of the chaperonin impurity. Protein Expression Purif. 2008, 60, 194–197. [Google Scholar] [CrossRef]
- Prive, G.G. Detergents for the stabilization and crystallization of membrane proteins. Methods 2007, 41, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Mazurkiewicz, P.; Driessen, A.J.; Konings, W.N. Energetics of wild-type and mutant multidrug resistance secondary transporter LmrP of Lactococcus lactis. Biochim. Biophys. Acta 2004, 1658, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Sigal, N.; Lewinson, O.; Wolf, S.G.; Bibi, E. E. coli multidrug transporter MdfA is a monomer. Biochemistry 2007, 46, 5200–5208. [Google Scholar] [CrossRef] [PubMed]
- Nobbmann, U.; Connah, M.; Fish, B.; Varley, P.; Gee, C.; Mulot, S.; Chen, J.; Zhou, L.; Lu, Y.; Shen, F.; et al. Dynamic light scattering as a relative tool for assessing the molecular integrity and stability of monoclonal antibodies. Biotechnol. Genet. Eng. Rev. 2007, 24, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Chae, P.S.; Cho, K.H.; Bae, H.E. Heavy atom-bearing tripod amphiphiles for the membrane protein study. N. J. Chem. 2014, 38, 2354–2361. [Google Scholar] [CrossRef]
- Lee, S.C.; Bennett, B.C.; Hong, W.X.; Fu, Y.; Baker, K.A.; Marcoux, J.; Robinson, C.V.; Ward, A.B.; Halpert, J.R.; Stevens, R.C.; et al. Steroid-based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Oliver, R.C.; Lipfert, J.; Fox, D.A.; Lo, R.H.; Doniach, S.; Columbus, L. Dependence of micelle size and shape on detergent alkyl chain length and head group. PLOS ONE 2013, 8, e62488. [Google Scholar] [CrossRef] [PubMed]
- Aivaliotis, M.; Samolis, P.; Neofotistou, E.; Remigy, H.; Rizos, A.K.; Tsiotis, G. Molecular size determination of a membrane protein in surfactants by light scattering. Biochim. Biophys. Acta 2003, 1615, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Calcutta, A.; Jessen, C.M.; Behrens, M.A.; Oliveira, C.L.; Renart, M.L.; Gonzalez-Ros, J.M.; Otzen, D.E.; Pedersen, J.S.; Malmendal, A.; Nielsen, N.C. Mapping of unfolding states of integral helical membrane proteins by GPS-NMR and scattering techniques: TFE-induced unfolding of KcsA in DDM surfactant. Biochim. Biophys. Acta 2012, 1818, 2290–2301. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Nachtergaele, S.; Seddon, A.M.; Tereshko, V.; Ponomarenko, N.; Ismagilov, R.F. Simple host-guest chemistry to modulate the process of concentration and crystallization of membrane proteins by detergent capture in a microfluidic device. J. Am. Chem. Soc. 2008, 130, 14324–14328. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, C. Multidrug Transporter MdfA as a Target for High-Resolution Structural Studies. Ph.D. Thesis, University of Saskatchewan, Saskatoon, SK, Canada, 2010. [Google Scholar]
- Boulter, J.M.; Wang, D.N. Purification and characterization of human erythrocyte glucose transporter in decylmaltoside detergent solution. Protein Expr. Purif. 2001, 22, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xia, D. The use of blue native page in the evaluation of membrane protein aggregation states for crystallization. J. Appl. Crystallogr. 2008, 41, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Newstead, S.; Hu, N.J.; Alguel, Y.; Nji, E.; Beis, K.; Yashiro, S.; Lee, C.; Leung, J.; Cameron, A.D.; et al. Benchmarking membrane protein detergent stability for improving throughput of high-resolution X-ray structures. Structure 2011, 19, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, M.J.; Reithmeier, R.A.; Wang, D.N. Importance of detergent and phospholipid in the crystallization of the human erythrocyte anion-exchanger membrane domain. J. Struct. Biol. 2002, 137, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Stroud, R.M. New tools in membrane protein determination. F1000 Biol. Rep. 2011. [Google Scholar] [CrossRef]
- Van de Weert, M. Fluorescence quenching to study protein-ligand binding: Common errors. J. Fluoresc. 2010, 20, 625–629. [Google Scholar]
- Pace, C.N.; Laurents, D.V. A new method for determining the heat capacity change for protein folding. Biochemistry 1989, 28, 2520–2525. [Google Scholar] [CrossRef] [PubMed]
- Handzlik, J.; Matys, A.; Kiec-Kononowicz, K. Recent advances in multi-drug resistance (MDR) efflux pump inhibitors of Gram-positive bacteria S. aureus. Antibiotics 2013, 2, 28–45. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alegre, K.O.; Law, C.J. Purification of a Multidrug Resistance Transporter for Crystallization Studies. Antibiotics 2015, 4, 113-135. https://doi.org/10.3390/antibiotics4010113
Alegre KO, Law CJ. Purification of a Multidrug Resistance Transporter for Crystallization Studies. Antibiotics. 2015; 4(1):113-135. https://doi.org/10.3390/antibiotics4010113
Chicago/Turabian StyleAlegre, Kamela O., and Christopher J. Law. 2015. "Purification of a Multidrug Resistance Transporter for Crystallization Studies" Antibiotics 4, no. 1: 113-135. https://doi.org/10.3390/antibiotics4010113
APA StyleAlegre, K. O., & Law, C. J. (2015). Purification of a Multidrug Resistance Transporter for Crystallization Studies. Antibiotics, 4(1), 113-135. https://doi.org/10.3390/antibiotics4010113