
Core Steps of Membrane-Bound Peptidoglycan Biosynthesis: Recent Advances, Insight and Opportunities

Abstract
:
1. Introduction

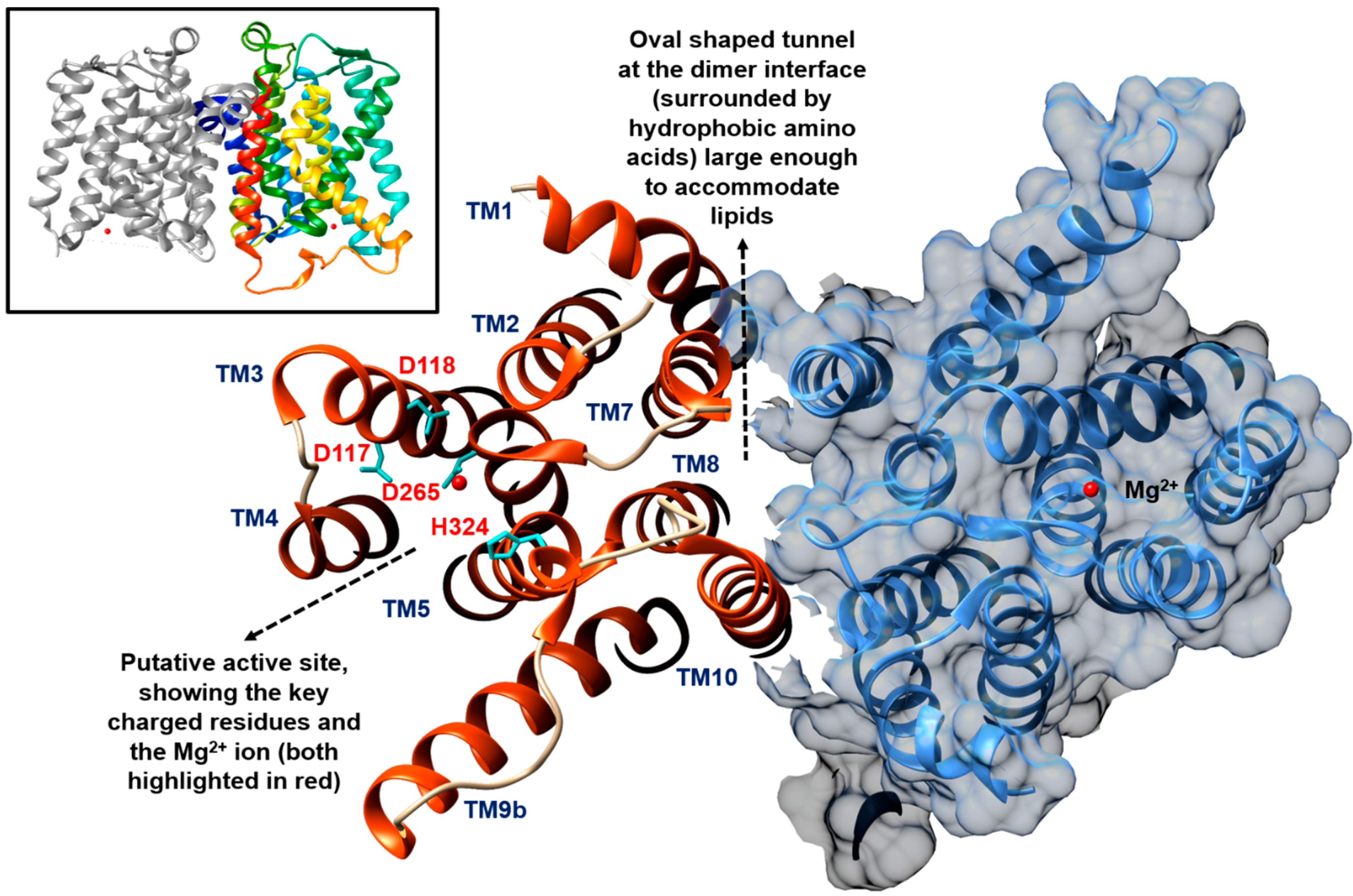
2. The Formation of Lipid I by MraY

3. The Formation of Lipid II by MurG

4. The Identity of the Lipid II Flippase
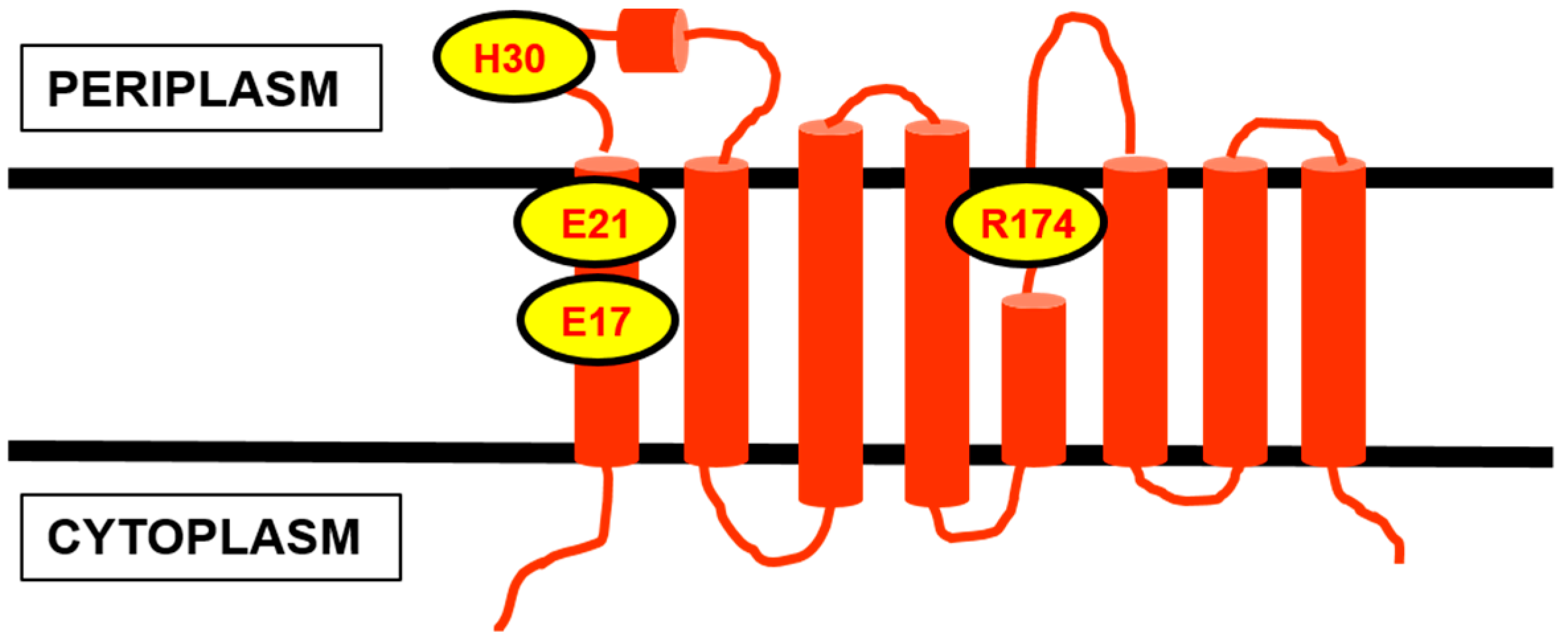
4.1. Contender I: FtsW
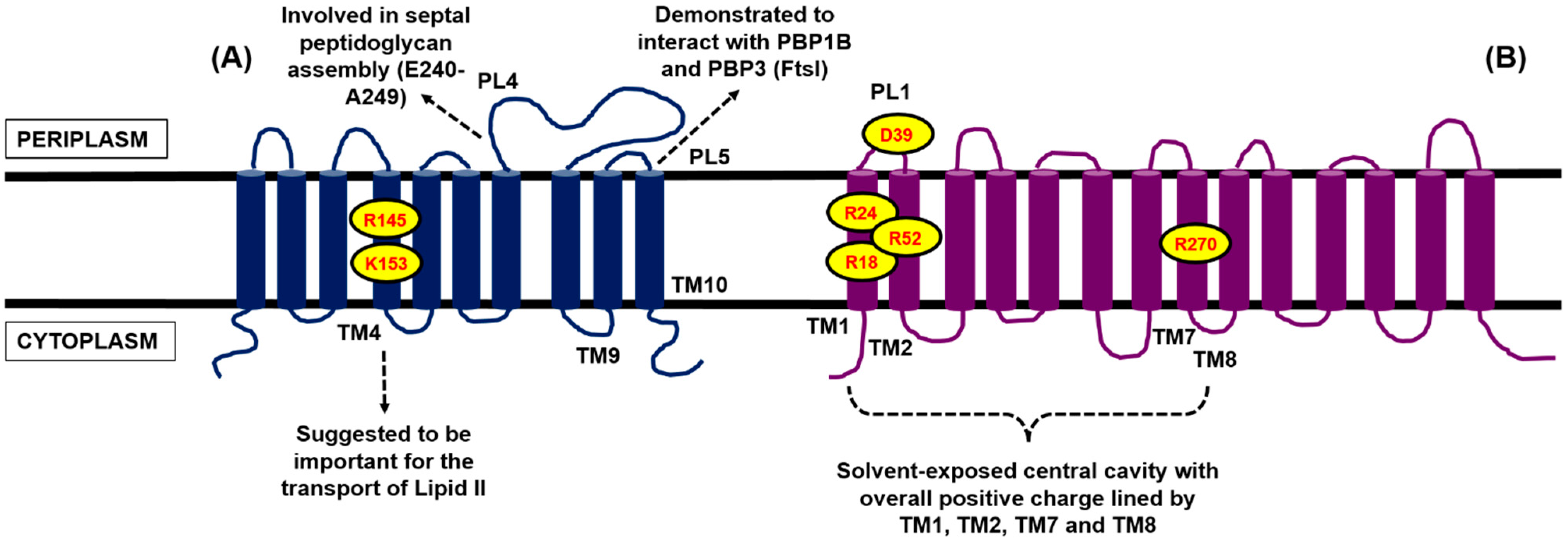
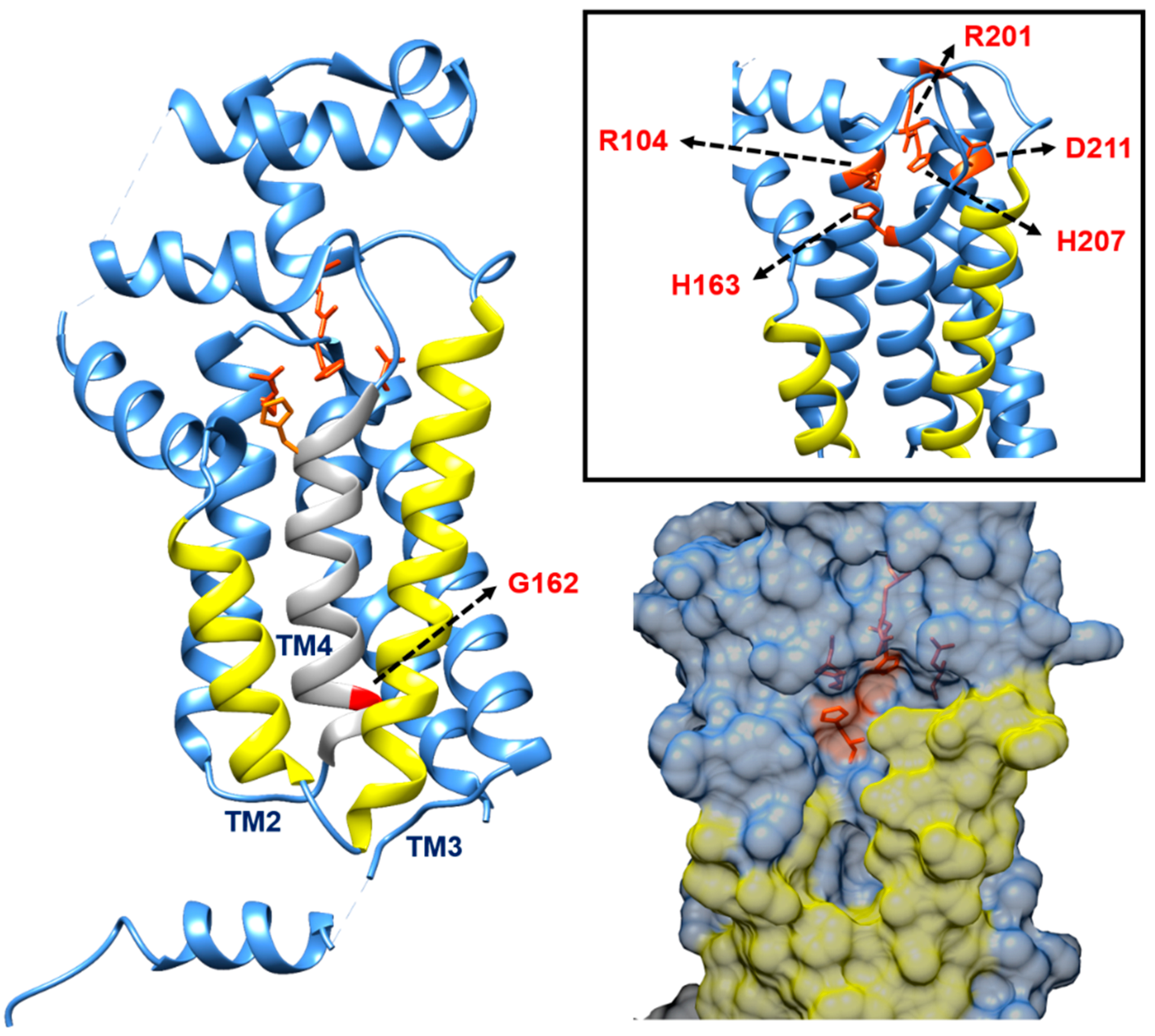
4.2. Contender II: MurJ

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Key Features |
|---|---|
| FtsW | |
| |
| MurJ | |
| |
| |
|
5. The Metabolism of C55-P: Multiple Enzymes Can Dephosphorylate C55-PP


| Protein | Key Features | |
|---|---|---|
| UppP/BacA |
| |
| ||
| ||
| PAP2 superfamily | PgpB | |
| ||
| YbjG |
| |
| ||
| ||
| YeiU/LpxT |
| |
| ||
| ||
| ||
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Antimicrobial Resistance: Global Report on Surveillance; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- O’Neill, J. Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. Available online: http://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (accessed on 1 July 2015).
- O’Neill, J. Review on Antimicrobial Resistance. Tackling a Global Health Crisis: Initial Steps. Available online: https://amr-review.org/sites/default/files/Report-52.15.pdf (accessed on 1 July 2015).
- Zapun, A.; Contreras-Martel, C.; Vernet, T. Penicillin-binding proteins and β-lactam resistance. FEMS Microbiol. Rev. 2008, 32, 361–385. [Google Scholar] [CrossRef] [PubMed]
- Typas, A.; Banzhaf, M.; Gross, C.A.; Vollmer, W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 2012, 10, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.L.; Safadi, S.S.; Strynadka, N.C.J. Structural perspective of peptidoglycan biosynthesis and assembly. Annu. Rev. Biochem. 2012, 81, 451–478. [Google Scholar] [CrossRef] [PubMed]
- Fuda, C.; Suvorov, M.; Vakulenko, S.B.; Mobashery, S. The basis for resistance to beta-lactam antibiotics by penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. J. Biol. Chem. 2004, 279, 40802–40806. [Google Scholar] [CrossRef] [PubMed]
- Dowson, C.G.; Hutchison, A.; Woodford, N.; Johnson, A.P.; George, R.C.; Spratt, B.G. Penicillin-resistant viridans streptococci have obtained altered penicillin-binding protein genes from penicillin-resistant strains of Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 1990, 87, 5858–5862. [Google Scholar] [CrossRef] [PubMed]
- Hakenbeck, R.; Brückner, R.; Denapaite, D.; Maurer, P. Molecular mechanisms of β-lactam resistance in Streptococcus pneumoniae. Future Microbiol. 2012, 7, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Tomberg, J.; Unemo, M.; Davies, C.; Nicholas, R.A. Molecular and structural analysis of mosaic variants of penicillin-binding protein 2 conferring decreased susceptibility to expanded-spectrum cephalosporins in Neisseria gonorrhoeae: Role of epistatic mutations. Biochemistry 2010, 49, 8062–8070. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Viable screening targets related to the bacterial cell wall. Ann. N. Y. Acad. Sci. 2013, 1277, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Does the cell wall of bacteria remain a viable source of targets for novel antibiotics? Biochem. Pharmacol. 2006, 71, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.H.; Braddick, D.; Dowson, C.G.; Roper, D.I. Bacterial cell wall assembly: Still an attractive antibacterial target. Trends Biotechnol. 2011, 29, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Barreteau, H.; Kovač, A.; Boniface, A.; Sova, M.; Gobec, S.; Blanot, D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 168–207. [Google Scholar] [CrossRef] [PubMed]
- Bouhss, A.; Trunkfield, A.E.; Bugg, T.D.H.; Mengin-Lecreulx, D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 2008, 32, 208–233. [Google Scholar] [CrossRef] [PubMed]
- van Heijenoort, J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev. 2007, 71, 620–635. [Google Scholar] [PubMed]
- Zapun, A.; Philippe, J.; Abrahams, K.A.; Signor, L.; Roper, D.I.; Breukink, E.; Vernet, T. In vitro reconstitution of peptidoglycan assembly from the gram-positive pathogen Streptococcus pneumoniae. ACS Chem. Biol. 2013, 8, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Matteï, P.J.; Neves, D.; Dessen, A. Bridging cell wall biosynthesis and bacterial morphogenesis. Curr. Opin. Struct. Biol. 2010, 20, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Bouhss, A.; Crouvoisier, M.; Blanot, D.; Mengin-lecreulx, D. Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan biosynthesis. J. Biol. Chem. 2004, 279, 29974–29980. [Google Scholar] [CrossRef] [PubMed]
- Price, N.P.; Momany, F.A. Modeling bacterial UDP-HexNAc: Polyprenol-P HexNAc-1-P transferases. Glycobiology 2005, 15, 29R–42R. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, J.; Vigeant, K.A.; Tatar, L.D.; Valvano, M.A. Functional characterization and membrane topology of Escherichia coli WecA, a sugar-phosphate transferase initiating the biosynthesis of enterobacterial common antigen and O-antigen lipopolysaccharide. J. Bacteriol. 2007, 189, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.J.; Brandish, P.E.; Gilbey, A.M.; Bugg, T.D.H. Phospho-N-acetyl-muramyl-pentapeptide translocase from Escherichia coli: Catalytic role of conserved aspartic acid residues. J. Bacteriol. 2004, 186, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Bouhss, A.; Mengin-Lecreulx, D.; Le Beller, D.; van Heijenoort, J. Topological analysis of the MraY protein catalysing the first membrane step of peptidoglycan synthesis. Mol. Microbiol. 1999, 34, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Al-Dabbagh, B.; Henry, X.; El Ghachi, M.; Auger, G.; Blanot, D.; Parquet, C.; Mengin-Lecreulx, D.; Bouhss, A. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry 2008, 47, 8919–8928. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Jahić, H.; Gao, N.; Hajec, L.; Rivin, O. A high-throughput, homogeneous, fluorescence resonance energy transfer-based assay for phospho-N-acetylmuramoyl-pentapeptide translocase (MraY). J. Biomol. Screen. 2012, 17, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Münch, D.; Schneider, T.; Sahl, H.-G.; Bouhss, A.; Ghoshdastider, U.; Wang, J.; Dötsch, V.; Wang, X.; Bernhard, F. Preparative scale cell-free production and quality optimization of MraY homologues in different expression modes. J. Biol. Chem. 2011, 286, 38844–38853. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.; Zocher, M.; Müller, D.; Münch, D.; Schneider, T.; Sahl, H.-G.; Scholz, F.; Wachtveitl, J.; Ma, Y.; Proverbio, D.; et al. Characterization of co-translationally formed nanodisc complexes with small multidrug transporters, proteorhodopsin and with the E. coli MraY translocase. Biochim. Biophys. Acta 2012, 1818, 3098–3106. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.C.; Zhao, J.; Gillespie, R.A.; Kwon, D.-Y.; Guan, Z.; Hong, J.; Zhou, P.; Lee, S.-Y. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science 2013, 341, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Heydanek, M.G.; Struve, W.G.; Neuhauss, F.C. On the initial stage in peptidoglycan synthesis. 3. Kinetics and uncoupling of phospho-N-acetylmuramyl-pentapeptide translocase (uridine 5′-phosphate). Biochemistry 1969, 8, 1214–1221. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Lloyd, A.J.; Roper, D.I. Phospho-MurNAc-pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect. Disord. Drug Targets 2006, 6, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Campbell, J.; Swoboda, J.G.; Cuny, G.D.; Walker, S. Development of improved inhibitors of wall teichoic acid biosynthesis with potent activity against Staphylococcus aureus. Bioorgan. Med. Chem. Lett. 2010, 20, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Simmons, K.J.; Chopra, I.; Fishwick, C.W.G. Structure-based discovery of antibacterial drugs. Nat. Rev. Microbiol. 2010, 8, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.; Goss, R.J.M.; Kimura, K.; Bugg, T.D.H. Antimicrobial nucleoside antibiotics targeting cell wall assembly: Recent advances in structure-function studies and nucleoside biosynthesis. Nat. Prod. Rep. 2010, 27, 279–304. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Zhang, W. Chemical logic and enzymatic machinery for biological assembly of peptidyl nucleoside antibiotics. ACS Chem. Biol. 2011, 6, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Engohang-Ndong, J. Antimycobacterial drugs currently in Phase II clinical trials and preclinical phase for tuberculosis treatment. Expert Opin. Investig. Drugs 2012, 21, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, Y.; Hayashi, C.; Inoue, K.; Igarashi, M.; Takahashi, Y.; Pujari, V.; Crick, D.C.; Brennan, P.J.; Nomoto, A. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J. Biol. Chem. 2013, 288, 30309–30319. [Google Scholar] [CrossRef] [PubMed]
- Mendel, S.; Holbourn, J.M.; Schouten, J.A.; Bugg, T.D.H. Interaction of the TM domain of lysis protein E from bacteriophage ϕX174 with bacterial translocase MraY and peptidyl-prolyl isomerase SlyD. Microbiology 2006, 152, 2959–2967. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, T.G.; Roof, W.D.; Young, R. Genetic evidence that the bacteriophage ΦX174 lysis protein inhibits cell wall synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 4297–4302. [Google Scholar] [CrossRef] [PubMed]
- Rodolis, M.T.; Mihalyi, A.; O’Reilly, A.; Slikas, J.; Roper, D.I.; Hancock, R.E.W.; Bugg, T.D.H. Identification of a novel inhibition site in translocase MraY based upon the site of interaction with lysis protein E from bacteriophage ϕX174. Chembiochem 2014, 15, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Walker, D.; Shi, Y.; Walker, S. The 1.9 A crystal structure of Escherichia coli MurG, a membrane-associated glycosyltransferase involved in peptidoglycan biosynthesis. Protein Sci. 2000, 9, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, L.; Ha, S.; Gross, B.; Falcone, B.; Walker, D.; Mokhtarzadeh, M.; Walker, S. Crystal structure of the MurG:UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proc. Natl. Acad. Sci. USA 2003, 100, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Gross, B.; Walker, S. E. coli MurG: A paradigm for a superfamily of glycosyltransferases. Curr. Drug Targets Infect. Disord. 2001, 1, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Vial, S.C.M.; Dedi, N.; Westcott, J.; Scally, S.; Bugg, T.D.H.; Charlton, P.A.; Cheetham, G.M.T. Crystal structure of the Pseudomonas aeruginosa MurG: UDP-GlcNAc substrate complex. Protein Pept. Lett. 2013, 20, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Crouvoisier, M.; Auger, G.; Blanot, D.; Mengin-Lecreulx, D. Role of the amino acid invariants in the active site of MurG as evaluated by site-directed mutagenesis. Biochimie 2007, 89, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Trunkfield, A.E.; Gurcha, S.S.; Besra, G.S.; Bugg, T.D.H. Inhibition of Escherichia coli glycosyltransferase MurG and Mycobacterium tuberculosis Gal transferase by uridine-linked transition state mimics. Bioorgan. Med. Chem. 2010, 18, 2651–2663. [Google Scholar] [CrossRef] [PubMed]
- Mann, P.A.; Müller, A.; Xiao, L.; Pereira, P.M.; Yang, C.; Lee, S.H.; Wang, H.; Trzeciak, J.; Schneeweis, J.; dos Santos, M.M.; et al. Murgocil is a highly bioactive Staphylococcal-specific inhibitor of the peptidoglycan glycosyltransferase enzyme MurG. ACS Chem. Biol. 2013, 8, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Pinho, M.G.; Errington, J. Recruitment of penicillin-binding protein PBP2 to the division site of Staphylococcus aureus is dependent on its transpeptidation substrates. Mol. Microbiol. 2005, 55, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Manat, G.; Roure, S.; Auger, R.; Bouhss, A.; Barreteau, H.; Mengin-Lecreulx, D.; Touzé, T. Deciphering the metabolism of undecaprenyl-phosphate: The bacterial cell-wall unit carrier at the membrane frontier. Microb. Drug Resist. 2014, 20, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, T.; van Dam, V.; Sijbrandi, R.; Vernet, T.; Zapun, A.; Bouhss, A.; Diepeveen-de Bruin, M.; Nguyen-Distèche, M.; de Kruijff, B.; Breukink, E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011, 30, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Lara, B.; Ayala, J.A. Topological characterization of the essential Escherichia coli cell division protein FtsW. FEMS Microbiol. Lett. 2002, 216, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gérard, P.; Vernet, T.; Zapun, A. Membrane topology of the Streptococcus pneumoniae FtsW division protein. J. Bacteriol. 2002, 184, 1925–1931. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.S.; Khattar, M.M.; Addinall, S.G.; Lutkenhaus, J.; Donachie, W.D. ftsW is an essential cell-division gene in Escherichia coli. Mol. Microbiol. 1997, 24, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Pastoret, S.; Fraipont, C.; den Blaauwen, T.; Wolf, B.; Aarsman, M.E.G.; Piette, A.; Thomas, A.; Brasseur, R.; Nguyen-Distèche, M. Functional analysis of the cell division protein FtsW of Escherichia coli. J. Bacteriol. 2004, 186, 8370–8379. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K.L.N.; Weiss, D.S. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J. Bacteriol. 2002, 184, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Fraipont, C.; Alexeeva, S.; Wolf, B.; van der Ploeg, R.; Schloesser, M.; den Blaauwen, T.; Nguyen-Distèche, M. The integral membrane FtsW protein and peptidoglycan synthase PBP3 form a subcomplex in Escherichia coli. Microbiology 2011, 157, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Butler, E.K.; Davis, R.M.; Bari, V.; Nicholson, P.A.; Ruiz, N. Structure-function analysis of MurJ reveals a solvent-exposed cavity containing residues essential for peptidoglycan biogenesis in Escherichia coli. J. Bacteriol. 2013, 195, 4639–4649. [Google Scholar] [CrossRef] [PubMed]
- van Dam, V.; Sijbrandi, R.; Kol, M.; Swiezewska, E.; de Kruijff, B.; Breukink, E. Transmembrane transport of peptidoglycan precursors across model and bacterial membranes. Mol. Microbiol. 2007, 64, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 15553–15557. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, T.; Sijbrandi, R.; Lutters, M.; Verheul, J.; Martin, N.; den Blaauwen, T.; de Kruijff, B.; Breukink, E. Specificity of the transport of Lipid II by FtsW in Escherichia coli. J. Biol. Chem. 2014, 289, 14707–14718. [Google Scholar] [CrossRef] [PubMed]
- Ehlert, K.; Höltje, J.-V. Role of precursor translocation in coordination of murein and phospholipid synthesis in Escherichia coli. J Bacteriol. 1996, 178, 6766–6771. [Google Scholar] [PubMed]
- Ikeda, M.; Sato, T.; Wachi, M.; Jung, H.K.; Ishino, F.; Kobayashi, Y.; Matsuhashi, M. Structural similarity among Escherichia coli FtsW and RodA proteins and Bacillus subtilis SpoVE protein, which function in cell division, cell elongation, and spore formation, respectively. J. Bacteriol. 1989, 171, 6375–6378. [Google Scholar] [PubMed]
- Hvorup, R.N.; Winnen, B.; Chang, A.B.; Jiang, Y.; Zhou, X.-F.; Saier, M.H., Jr. The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur. J. Biochem. 2003, 270, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Murata, Y.; Takahashi, H.; Tsuji, N.; Fujisaki, S.; Kato, J. Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J. Bacteriol. 2008, 190, 7298–7301. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, Y.F.; Valvano, M.A. A Burkholderia cenocepacia MurJ (MviN) homolog is essential for cell wall peptidoglycan synthesis and bacterial viability. Glycobiology 2014, 24, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Butler, E.K.; Tan, W.B.; Joseph, H.; Ruiz, N. Charge requirements of Lipid II flippase activity in Escherichia coli. J. Bacteriol. 2014, 196, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, N. Streptococcus pyogenes YtgP (Spy_0390) complements Escherichia coli strains depleted of the putative peptidoglycan flippase MurJ. Antimicrob. Agents Chemother. 2009, 53, 3604–3605. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.-T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Fay, A.; Dworkin, J. Bacillus subtilis homologs of MviN (MurJ), the putative Escherichia coli lipid II flippase, are not essential for growth. J. Bacteriol. 2009, 191, 6020–6028. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, P.; McElligott, J.; Attkisson, C.; Betteken, M.; Popham, D.L. Homologues of the Bacillus subtilis SpoVB protein are involved in cell wall metabolism. J. Bacteriol. 2009, 191, 6012–6019. [Google Scholar] [CrossRef] [PubMed]
- Meeske, A.J.; Sham, L.-T.; Kimsey, H.; Koo, B.-M.; Gross, C.A.; Bernhardt, T.G.; Rudner, D.Z. MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2015, 112, 6437–6442. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.; Donald, R.G.K.; Lee, S.H.; Jarantow, L.W.; Salvatore, M.J.; Meng, X.; Painter, R.; Onishi, R.H.; Occi, J.; Dorso, K.; et al. Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem. Biol. 2009, 16, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Fujihashi, M.; Zhang, Y.-W.; Higuchi, Y.; Li, X.-Y.; Koyama, T.; Miki, K. Crystal structure of cis-prenyl chain elongating enzyme, undecaprenyl diphosphate synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 4337–4342. [Google Scholar] [CrossRef] [PubMed]
- El Ghachi, M.; Bouhss, A.; Blanot, D.; Mengin-Lecreulx, D. The bacA gene of Escherichia coli encodes an undecaprenyl pyrophosphate phosphatase activity. J. Biol. Chem. 2004, 279, 30106–30113. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-Y.; Chou, C.-C.; Hsu, M.-F.; Wang, A.H.J. Proposed carrier lipid-binding site of undecaprenyl pyrophosphate phosphatase from Escherichia coli. J. Biol. Chem. 2014, 289, 18719–18735. [Google Scholar] [CrossRef] [PubMed]
- Tatar, L.D.; Marolda, C.L.; Polischuk, A.N.; van Leeuwen, D.; Valvano, M.A. An Escherichia coli undecaprenyl-pyrophosphate phosphatase implicated in undecaprenyl phosphate recycling. Microbiology 2007, 153, 2518–2529. [Google Scholar] [CrossRef] [PubMed]
- Bickford, J.S.; Nick, H.S. Conservation of the PTEN catalytic motif in the bacterial undecaprenyl pyrophosphate phosphatase, BacA/UppP. Microbiology 2013, 159, 2444–2455. [Google Scholar] [CrossRef] [PubMed]
- Bernard, R.; El Ghachi, M.; Mengin-Lecreulx, D.; Chippaux, M.; Denizot, F. BcrC from Bacillus subtilis acts as an undecaprenyl pyrophosphate phosphatase in bacitracin resistance. J. Biol. Chem. 2005, 280, 28852–28857. [Google Scholar] [CrossRef] [PubMed]
- El Ghachi, M.; Derbise, A.; Bouhss, A.; Mengin-Lecreulx, D. Identification of multiple genes encoding membrane proteins with undecaprenyl pyrophosphate phosphatase (UppP) activity in Escherichia coli. J. Biol. Chem. 2005, 280, 18689–18695. [Google Scholar] [CrossRef] [PubMed]
- Shaaly, A.; Kalamorz, F.; Gebhard, S.; Cook, G.M. Undecaprenyl pyrophosphate phosphatase confers low-level resistance to bacitracin in Enterococcus faecalis. J. Antimicrob. Chemother. 2013, 68, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Kjos, M.; Oppegård, C.; Diep, D.B.; Nes, I.F.; Veening, J.-W.; Nissen-Meyer, J.; Kristensen, T. Sensitivity to the two-peptide bacteriocin lactococcin G is dependent on UppP, an enzyme involved in cell-wall synthesis. Mol. Microbiol. 2014, 92, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Touzé, T.; Blanot, D.; Mengin-lecreulx, D. Substrate specificity and membrane topology of Escherichia coli PgpB, an undecaprenyl pyrophosphate phosphatase. J. Biol. Chem. 2008, 283, 16573–16583. [Google Scholar] [CrossRef] [PubMed]
- Valvano, M.A. Undecaprenyl phosphate recycling comes out of age. Mol. Microbiol. 2008, 67, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Touzé, T.; Tran, A.X.; Hankins, J.V.; Mengin-Lecreulx, D.; Trent, M.S. Periplasmic phosphorylation of lipid A is linked to the synthesis of undecaprenyl phosphate. Mol. Microbiol. 2008, 67, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Hynninen, A.; Touzé, T.; Pitkänen, L.; Mengin-Lecreulx, D.; Virta, M. An efflux transporter PbrA and a phosphatase PbrB cooperate in a lead-resistance mechanism in bacteria. Mol. Microbiol. 2009, 74, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Stukey, J.; Carman, G.M. Identification of a novel phosphatase sequence motif. Protein Sci. 1997, 6, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Icho, T.; Raetz, C.R.H. Multiple genes for membrane-bound phosphatases in Escherichia coli and their action on phospholipid precursors. J. Bacteriol. 1983, 153, 722–730. [Google Scholar] [PubMed]
- Icho, T. Membrane-bound phosphatases in Escherichia coli: Sequence of the pgpB gene and dual subcellular localization of the pgpB product. J. Bacteriol. 1988, 170, 5117–5124. [Google Scholar] [PubMed]
- Funk, C.R.; Zimniak, L.; Dowhan, W. The pgpA and pgpB genes of Escherichia coli are not essential: Evidence for a third phosphatidylglycerophosphate phosphatase. J. Bacteriol. 1992, 174, 205–213. [Google Scholar] [PubMed]
- Dillon, D.A.; Wu, W.-I.; Riedel, B.; Wissing, J.B.; Dowhan, W.; Carman, G.M. The Escherichia coli pgpB gene encodes for a diacylglycerol pyrophosphate phosphatase activity. J. Biol. Chem. 1996, 271, 30548–30553. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Jiang, D.; Zhao, Y.; Liu, J.; Zhang, X.C. Crystal structure of lipid phosphatase Escherichia coli phosphatidylglycerophosphate phosphatase B. Proc. Natl. Acad. Sci. USA 2014, 111, 7636–7640. [Google Scholar] [CrossRef] [PubMed]
- Young, K.D. A flipping cell wall ferry. Science 2014, 345, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Burda, P.; Aebi, M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1999, 1426, 239–257. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teo, A.C.K.; Roper, D.I. Core Steps of Membrane-Bound Peptidoglycan Biosynthesis: Recent Advances, Insight and Opportunities. Antibiotics 2015, 4, 495-520. https://doi.org/10.3390/antibiotics4040495
Teo ACK, Roper DI. Core Steps of Membrane-Bound Peptidoglycan Biosynthesis: Recent Advances, Insight and Opportunities. Antibiotics. 2015; 4(4):495-520. https://doi.org/10.3390/antibiotics4040495
Chicago/Turabian StyleTeo, Alvin C. K., and David I. Roper. 2015. "Core Steps of Membrane-Bound Peptidoglycan Biosynthesis: Recent Advances, Insight and Opportunities" Antibiotics 4, no. 4: 495-520. https://doi.org/10.3390/antibiotics4040495
APA StyleTeo, A. C. K., & Roper, D. I. (2015). Core Steps of Membrane-Bound Peptidoglycan Biosynthesis: Recent Advances, Insight and Opportunities. Antibiotics, 4(4), 495-520. https://doi.org/10.3390/antibiotics4040495