1. Introduction
Colicins are bacterial toxins produced by some
Escherichia coli strains in order to kill susceptible strains of
E. coli and related species. Among the ca
. twenty colicins identified to date [
1], colicin M (ColM) is unique as it is the only one interfering with bacterial peptidoglycan biosynthesis. ColM acts by cleavage of the last precursor of this essential metabolic pathway, thereby causing cell death. Indeed, it was shown ten years ago, both in vivo and in vitro, that ColM was an enzyme catalyzing specifically the hydrolysis of the peptidoglycan lipid intermediate C
55-PP-MurNAc(pentapeptide)-GlcNAc (lipid II), by cleaving between the prenyl chain (C
55-) and the pyrophosphoryl group of this precursor [
2]. The two reaction products, undecaprenol and 1-PP-MurNAc(pentapeptide)-GlcNAc, that do not normally exist in
E. coli growing cells, were accumulated in the ColM-treated cells. These products cannot be reused into peptidoglycan metabolism or be recycled, thereby leading to cell lysis.
ColM develops its bacteriolytic activity in three sequential steps deeply linked to its structural organization in three domains and parasitizes proteins from the targeted cell to enter the periplasm. Accordingly, the ColM central domain first binds to an outer membrane receptor for siderophores, FhuA [
3]. Subsequently, it is translocated towards the periplasm by using the proton-motive force, through the interaction of its N-terminal domain with the TonB machinery system comprising the TonB, ExbB and ExbD proteins [
4]. Finally, once shuttled into the periplasm of the targeted cell, ColM is maturated by the ubiquitous chaperone protein FkpA, so that its C-terminal domain can display its cytotoxic effect on lipid II [
5,
6].
As this is the case for many bacteriocins, ColM-producing cells are protected against the lethal effect of their own colicin by the concomitant synthesis of an immunity protein, named ImM (or Cmi). Although the latter protein has been structurally characterized [
7,
8], the mechanism of protection of the bacterial cell from lysis is still unknown. However, non-producing cells can also protect themselves against the action of ColM by mutation or deletion of genes encoding proteins involved in ColM reception/translocation/maturation processes [
9,
10].
During the last few years, several ColM orthologues have been identified, by sequence alignments of their C-terminal domains, in some other bacterial genera, such as
Pseudomonas,
Burkholderia and
Pectobacterium species, and several of them have been both biochemically and structurally characterized [
11,
12,
13,
14,
15]. All of the purified ColM orthologues displayed the same enzymatic activity of cleavage of lipid II and a bacteriolytic (or at least bacteriostatic) activity [
11,
12]. Yet, despite their similarities in terms of enzymatic activity, these orthologues did not share the same 3D structure, especially in their N-terminal domains. On the one hand, ColM shared a very compact structure with PaeM and syringacin M (also named PsyM), i.e., its orthologues from
Pseudomonas aeruginosa and
Pseudomonas syringae, respectively, where the usual three structural domains did not form distinct entities [
12,
13]. On the other hand, the structure of pectocin M2 from
Pectobacterium carotovorum and probably also that of pectocin M1, since it has been strongly suggested that both pectocins M1 and M2 used the same outer membrane receptor on
Pectobacterium spp. [
14], presented an atypical translocation domain, where the helical receptor-domain and unstructured N-terminus were replaced by a single globular plant-like [2Fe-2S] ferredoxin domain, directly connected to the cytotoxic one through a small α-helix linker [
15]. Moreover, while numerous receptors for colicins and closely-related pyocins were Tol- or TonB-dependent, these ferredoxin-containing pectocins presumably used a bacterial ferredoxin uptake mechanism to cross the outer membrane, without any evidence for Tol or TonB complex requirements [
15].
The cytotoxicity of all of these ColM-like bacteriocins has been previously demonstrated to be pointed towards a limited number of bacterial species. The specificity of their receptor-binding and translocation domains presumably prevents them from targeting a broad range of bacteria. It thus seems that, if reaching the target constitutes a crucial step for the colicin cytotoxicity, it is also the limiting one. Yet, ways allowing the bypass of this specific and limiting step do exist. Indeed, the use of an osmotic shock was shown to be efficient to make extracellular proteins enter the periplasm of Gram-negative bacteria without loss of cell integrity [
16,
17]. Another efficient means to send proteins into the periplasm of a target cell was the use of fusion proteins between the bacteriocin of interest and a signal sequence. This strategy was particularly used to study the pore formation by colicin A [
18,
19]. According to the latter methodology, we describe in the present paper how we managed to evaluate the antibacterial activity of pectocin M1 (that will be named PcaM1 throughout this work, following the nomenclature we previously used [
11]) and some of its variants on
E. coli cells by triggering their periplasmic expression. We show that when addressed to the periplasm of
E. coli, the PcaM1 protein, as well as its isolated activity domain could catalyze the degradation of the peptidoglycan lipid II precursor, which leads to the arrest of the biosynthesis of this essential cell wall component and, consequently, to cell lysis. It is also shown that, contrary to ColM, this pectocin does not depend on the presence of the host FkpA chaperone protein to exert its deleterious toxic effects.
3. Discussion
In this work, we focused our investigations on PcaM1, one of the two ColM-like orthologues produced by P. carotovorum. We first revisited the results previously published by Grinter and collaborators in 2012 by producing the wild-type and catalytic point mutant D222A PcaM1 proteins. We thus confirmed, on the one hand, the enzymatic activity of lipid II degradation by the wild-type PcaM1, whereas the mutant was inactive, and on the other hand, the absence of cytotoxic activity of PcaM1 preparation on E. coli cells, as no growth inhibition zone was detected when up to 57 µg of this protein were spotted on E. coli BW25113-inoculated agar plates.
Although PcaM1, as well as the other ColM orthologues [
11] did not display any cytotoxic activity against
E. coli cells, we recently showed that the application of an osmotic shock treatment to
E. coli cells allowed the
P. aeruginosa ColM orthologue (PaeM) to bypass the outer membrane reception and translocation steps, reach its lipid II target and thus exert its deleterious activity [
13]. This allowed us to demonstrate that PaeM, as well as its isolated catalytic domain, was able to kill
E. coli cells. It was the first example of a ColM-like protein capable of killing another bacterial species.
Another way to get access to the
E. coli periplasmic space was to fuse ColM to the OmpA protein signal sequence. In these conditions, the hybrid ColM was directly exported from the cytoplasm to the periplasm of the producing cells and was then demonstrated to be toxic [
22]. Therefore, to check whether another ColM orthologue would be able to kill
E. coli cells by this approach, we fused the PcaM1 to OmpA signal sequence and expressed the hybrid protein in
E. coli. In this way, we visualized that PcaM1 was toxic for
E. coli cells, as the controlled induction of its expression led to cell lysis. To the best of our knowledge, this is the first example of cell lysis due to periplasmic expression of a ColM-like orthologue. A hybrid protein obtained by fusion of the PcaM1 variant truncated of its 107 first amino acids to the OmpA signal sequence led to the same lytic phenotype, as also did the D222A point mutant PcaM1. That the expression of the isolated catalytic domain of PcaM1 yielded cell lysis in these conditions was somewhat expected, as this had been observed also with the catalytic domain of ColM [
22]. However, the lytic effect exhibited by the D222A variant was much more surprising. Indeed, as described above, no enzymatic activity of in vitro degradation of lipid II was reported for this mutant protein. The Asp222 residue from PcaM1 corresponds to Asp226 in ColM, which has been previously demonstrated to be essential for the catalytic activity and, consequently, for the cytotoxicity of ColM [
21]. Moreover, it was previously shown that the D226N mutant of ColM did not display any toxic activity when exported to the periplasm [
22]. The lytic phenotype of the
E. coli strain expressing the D222A PcaM1 mutant was thus intriguing. Cell lysis was observed even at low doses of anhydrotetracycline, suggesting that toxicity is due to an effect of mutant PcaM1 per se and not to a potential toxicity of protein overexpressed (
Figures S3 and S4).
As the induction of periplasmic PcaM1 expression triggered
E. coli cell lysis whatever the PcaM1 variant expressed, we investigated whether this phenotype was indeed the consequence of an arrest of peptidoglycan biosynthesis. Accordingly, ColM and its orthologue PaeM were previously shown to exhibit bacteriolytic and bacteriostatic effects on their specific
E. coli and
Pseudomonas targeted species, respectively, that were in both cases correlated to an inhibition of peptidoglycan biosynthesis [
2,
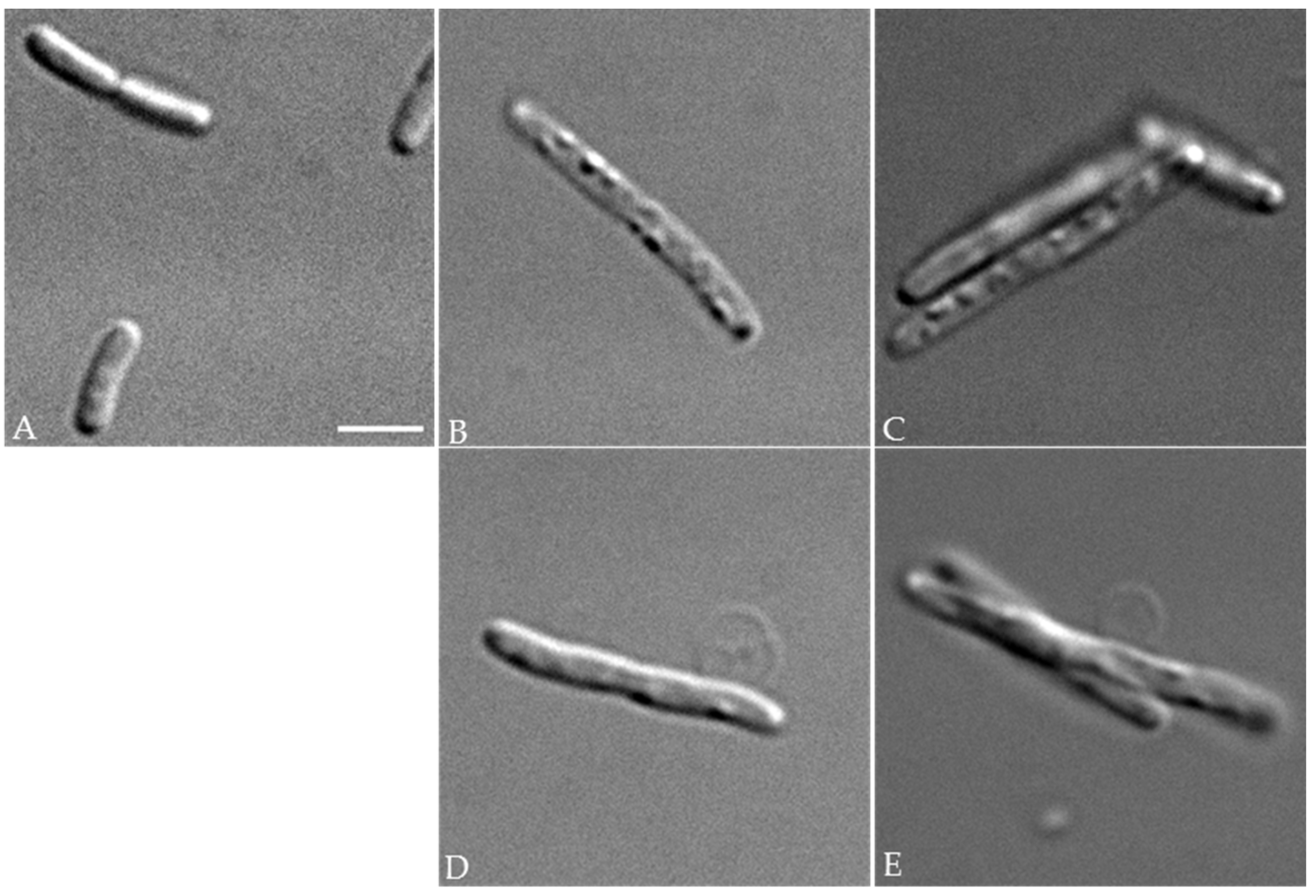
11]. In this respect, optical microscopy analyses showed that PcaM1-producing cells displayed greatly altered, elongated and bloated morphologies that are characteristic of an impaired cell wall biogenesis. The arrest of peptidoglycan synthesis in these cells was clearly demonstrated by (i) radiolabeling experiments, using
meso-[
14C]A
2pm as a specific marker, that revealed the accumulation of the cytoplasmic UDP-MurNAc-pentapeptide precursor and the concomitant arrest of the synthesis of the polymer, as well as the presence of the lipid II degradation product; (ii) analyses of isoprenoid pool levels in membranes, which revealed the appearance and accumulation of C
55-OH, which normally do not exist in
E. coli cells. Quite interestingly, although the D222A PcaM1 mutant did not show detectable lipid II-degrading enzymatic activity, either in vitro (enzymatic assays) or in vivo (no C
55-OH or 1-PP-MurNAc(pentapeptide)-GlcNAc detected in the cell content), its expression in the periplasm led to the same and specific dramatic morphological changes and arrest of peptidoglycan synthesis that have been observed with the functional wild-type PcaM1. This intriguing result could be interpreted in several ways. For instance, the D222A mutant whose lipid II hydrolase activity is abolished likely conserved its ability to interact with this substrate. A sequestration of the lipid II by the mutant protein could thus be envisaged, which would result in an inhibition of peptidoglycan polymerization steps catalyzed by the penicillin-binding proteins (PBP). Validation of such a hypothesis would need to precisely determine the relative numbers of lipid II and PcaM1 molecules present in the periplasmic space of
E. coli in these conditions, as well as to develop in vitro analyses of these protein-substrate interactions. Further work is thus needed to elucidate by which mechanism this inactive variant of PcaM1 could still interfere so efficiently with the peptidoglycan synthesis pathway.
In this study, we also tested the periplasmic expression of the three PcaM1 variants in an
E. coli strain carrying a deletion of the FkpA chaperone-encoding gene. Contrary to what was previously observed with periplasmic variants of ColM, i.e., that the full-length ColM protein and its isolated catalytic domain were FkpA-dependent and -independent, respectively [
22], all of the PcaM1 variants constructed here were able to induce
E. coli cell lysis in the absence of FkpA, meaning that no maturation process, at least by this chaperone, was needed for them to be toxic in
E. coli. It thus seems that the PcaM1 protein was produced right away in an active form in these particular conditions of heterologous expression.
This work thus clearly demonstrates that the limiting reception and translocation steps usually required for colicin cytotoxic activity can be bypassed. We already knew that ColM-like orthologues were able to hydrolyze in vitro peptidoglycan lipid II intermediates of various composition and structure originating from major pathogenic bacteria (
Staphylococcus aureus,
Enterococcus faecium, E. faecalis) [
25], and we confirmed that they could potentially do it also in vivo, as soon as they can reach the periplasmic space of the targeted bacterial species. In
E. coli, the ColM immunity protein, Cmi (or ImM), that prevents ColM toxicity effect is located in this compartment. A gene coding for a Cmi homologue has been identified in the genome of
P. carotovorum PC1, which likely confers immunity towards PcaM1 in this species [
14]. As these enzymatic colicins clearly constitute powerful molecules with great potential as non-conventional antimicrobial agents, we now consider engineering them in order to design chimera colicins able to exhibit a broader spectrum of antibacterial activity.
4. Materials and Methods
4.1. Bacterial Strains, Plasmids and Growth Conditions
The
E. coli strains DH5α (Bethesda Research Laboratories) and C43(DE3) (Avidis) were used as the hosts for the propagation of plasmids and the production of proteins, respectively. The
E. coli strains FB8 and BW25113 were used for bacteriolytic activity assays, while the
E. coli FB8 Δ
lysA::kan was used for
meso-[
14C]A
2pm incorporation experiments [
24]. The
Pectobacterium carotovorum ssp
. carotovorum strain PC1 was kindly provided by Dr. Iris Yedidia [
26]. The construction of the plasmid vector pET2160, a pET21d derivative allowing the expression of proteins with a C-terminal 6× histidine tag (His
6), has been previously described [
11]. The pREP4groESL plasmid allowing overexpression of the bacterial chaperones was obtained from Amrein, K., et al. [
27]. The pASK-IBA4 vector was used for the export of proteins to the periplasm of
E. coli, as described previously [
22]. For cloning experiments, protein production and lysis experiments, cells were grown aerobically at 37 °C in 2YT medium [
28], whereas they were grown in M63 minimum medium supplemented with 0.4% glucose and 100 µg/mL each of lysine, threonine and methionine for
meso-[
14C]A
2pm incorporation experiments [
2,
24]. When needed, ampicillin and kanamycin were used at 100 and 50 µg/mL, respectively. Growth was monitored at 600 nm with a Shimadzu UV-1601 spectrophotometer.
4.2. Molecular Biology Techniques
Polymerase chain reaction (PCR) amplification of genes was performed in a Thermocycler 60 apparatus (Bio-Med, Guilford, CT, USA) using the Expand-Fidelity polymerase (Roche Applied Science, Indianapolis, IN, USA). DNA fragments were purified with the Wizard PCR Preps DNA purification kit (Promega, Charbonnières-les-Bains, France), and standard procedures for DNA digestion, ligation, agarose gel electrophoresis and plasmid isolations were used [
29].
E. coli cells were transformed with plasmid DNA by the method of Dagert and Ehrlich [
30] or by electroporation.
4.3. Construction of Expression Plasmids
A plasmid allowing high-level overproduction of the ColM homologue gene from
P. carotovorum (
pcaM) was constructed as follows: PCR primers Pcam-O1 and Pcam-O2 (
Table 3) were designed to incorporate NcoI and BglII sites at the 5′ and 3′ extremities of the gene, respectively. The gene was amplified from the PC1 strain chromosome, and the DNA fragment was treated with NcoI and BglII and ligated between the same sites of the vector pET2160. The resulting plasmid, pMLD365, allowed the expression of the protein with a His
6-tag (Arg-Ser-His
6 extension) at the C-terminal extremity. To produce the D222A catalytic point mutant of PcaM1, site-directed mutagenesis of the C-terminal His
6-tagged protein was performed directly on the pMLD365 expression plasmid by using the QuikChange II XL mutagenesis kit (Stratagene, La Jolla, CA, USA), using the pair of complementary nucleotides Pcam-mut1 and Pcam-mut2. This yielded the pMLD464 plasmid.
Export of the PcaM protein to the periplasm of E. coli was obtained by fusing the sequence of this bacteriocin to that of the signal peptide of OmpA, using the pASK-IBA4 plasmid as the vector. The gene was amplified with Pcam-O1 and Pcam-O3 primers, and the fragment was cleaved by NcoI and HindIII and inserted between the same sites of pASK-IBA4, yielding the pMLD381 plasmid. Plasmid pMLD395, a pMLD381 derivative plasmid expressing the D222A PcaM1 mutant, was generated by site-directed mutagenesis using the Pcam-mut1 and Pcam-mut2 oligonucleotides. Plasmid pMLD403, a pASK-IBA4 derivative plasmid expressing an N-terminally-truncated PcaM1 variant lacking the first 107 residues, was generated as described for pMLD381, except that the oligonucleotides used for PCR gene amplification were the Pcam-Δ1-107 and Pcam-O3 primers.
4.4. Production and Purification of Wild-Type and Mutant Forms of PcaM1
For the expression of wild-type and mutant PcaM1 proteins, C43(DE3) cells were transformed with the pMLD365 or pMLD464 plasmid, respectively, as well as with the chaperone-expressing plasmid pREP4groESL. Cells were grown at 37 °C in 2YT medium supplemented with ampicillin and kanamycin (1-liter cultures), and when the optical density at 600 nm (OD600) of the culture reached 0.8, isopropyl-β-d-thiogalactopyranoside (IPTG) was added at a final concentration of 1 mM and growth continued at 22 °C overnight. Then, the cells were harvested, washed with 40 mL of an 0.9% NaCl solution and finally suspended in 12 mL of Buffer A (20 mM Tris-HCl, pH 7.2, 200 mM NaCl, 0.1% 2-mercaptoethanol, 0.5 mM MgCl2 and 10% glycerol). In each case, the bacterial suspension was disrupted by sonication (Bioblock Vibracell sonicator, model 72412, Fisher Scientific, Illkirch, France) and then centrifuged at 4 °C for 30 min at 200,000× g in a TL100 Beckman centrifuge. The wild-type or mutant PcaM1-containing supernatant was subjected to purification.
His6-tagged wild-type and mutant forms of PcaM1 were purified first by affinity chromatography on nickel-nitrilotriacetate (Ni2+-NTA)-agarose polymer (Qiagen®, Courtaboeuf, France). All procedures were performed at 4 °C. The crude soluble supernatant obtained according to the procedure described above was mixed with 2 mL of polymer pre-equilibrated with Buffer A containing 10 mM imidazole and incubated for 30 min at 4 °C according to the Qiagen® recommendations. Then, the washing and elution steps were performed with a discontinuous gradient of imidazole (20–200 mM) in Buffer A. Eluted proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The relevant fractions were pooled, concentrated to a volume of 5 mL by centrifugation on a 10-kDa cut-off membrane (Amicon Ultra, Millipore, Molsheim, France) and then submitted to an extra purification step by gel filtration (Äkta Prime system, ©GE Healthcare, Buckinghamshire, UK) on a Hi-Load 16/600 Superdex S200 column (©GE) pre-equilibrated with one column volume of Buffer A without MgCl2 and glycerol and previously calibrated with blue dextran, conalbumin, ovalbumin, carbonic anhydrase, ribonuclease, aprotinine and tyrosine. Elution was performed with the same buffer at a flow rate of 1 mL/min. The purity of the fractions corresponding to the PcaM1 elution peak was checked by SDS-PAGE, and the final protein concentration was determined by using a NanoDropTM 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The absorption spectra of purified wild-type and mutant forms of PcaM1 were determined using a Jasco V-630 spectrophotometer. Glycerol (10% v/v, final concentration) was eventually added for storage of the protein at −20 °C.
4.5. Hydrolase Activity Assays
The PcaM1 enzymatic activity was tested in a reaction mixture (10 µL) containing 100 mM Tris-HCl, pH 7.5, 20 mM MgCl
2, 150 mM NaCl, 10 mM 2-mercaptoethanol, 12 µM [
14C]radiolabeled lipid II (140 Bq) and 0.2%
n-dodecyl-β-
d-maltopyranoside (DDM), as previously described [
11]. The reaction was started by the addition of the purified protein (in 5 µL of Buffer A) and incubated for 30 min at 37 °C with shaking (Thermomixer, Eppendorf, Wesseling-Berzdorf, Germany). For the determination of the Michaelis constants (
Km), assay conditions were as described above, except that the concentration of lipid II varied from 6–100 µM. The reaction was stopped by heating at 95 °C for 1 min, and mixtures were analyzed by thin-layer chromatography (TLC) on pre-coated silica gel 60 F
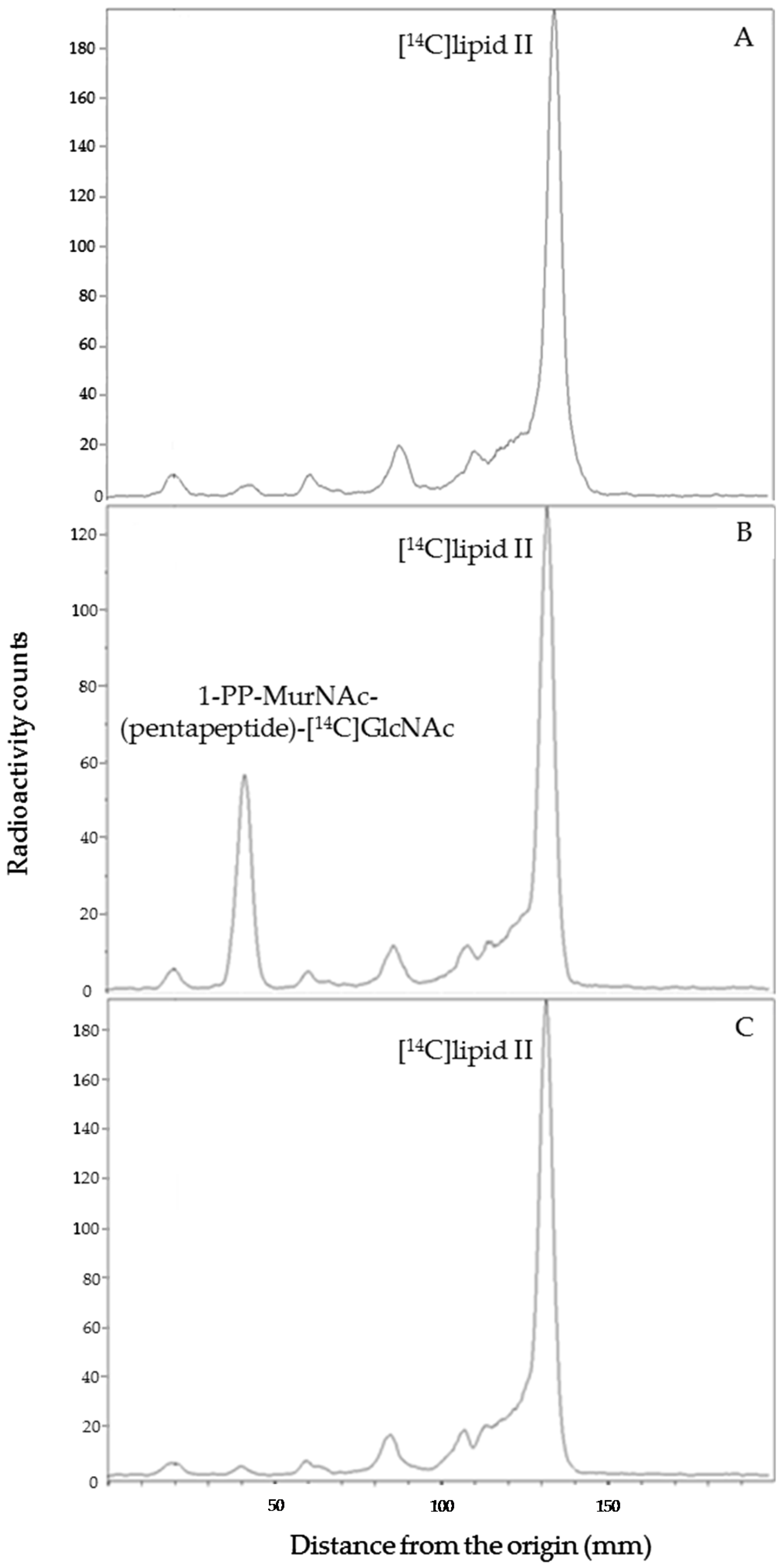
254 plates (Merck, Molsheim, France) using 1-propanol/ammonium hydroxide/water (6:3:1; v/v/v) as the mobile phase. The radioactive spots corresponding to the substrate (lipid II) and product (1-PP-MurNAc [pentapeptide]-GlcNAc) were located (
Rf = 0.7 and 0.3, respectively) and quantified with a radioactivity scanner (Rita Star, Raytest Isotopenmeßgeräte GmbH, Straubenhardt, Germany).
4.6. Bacteriolytic Activity Assays on E. coli
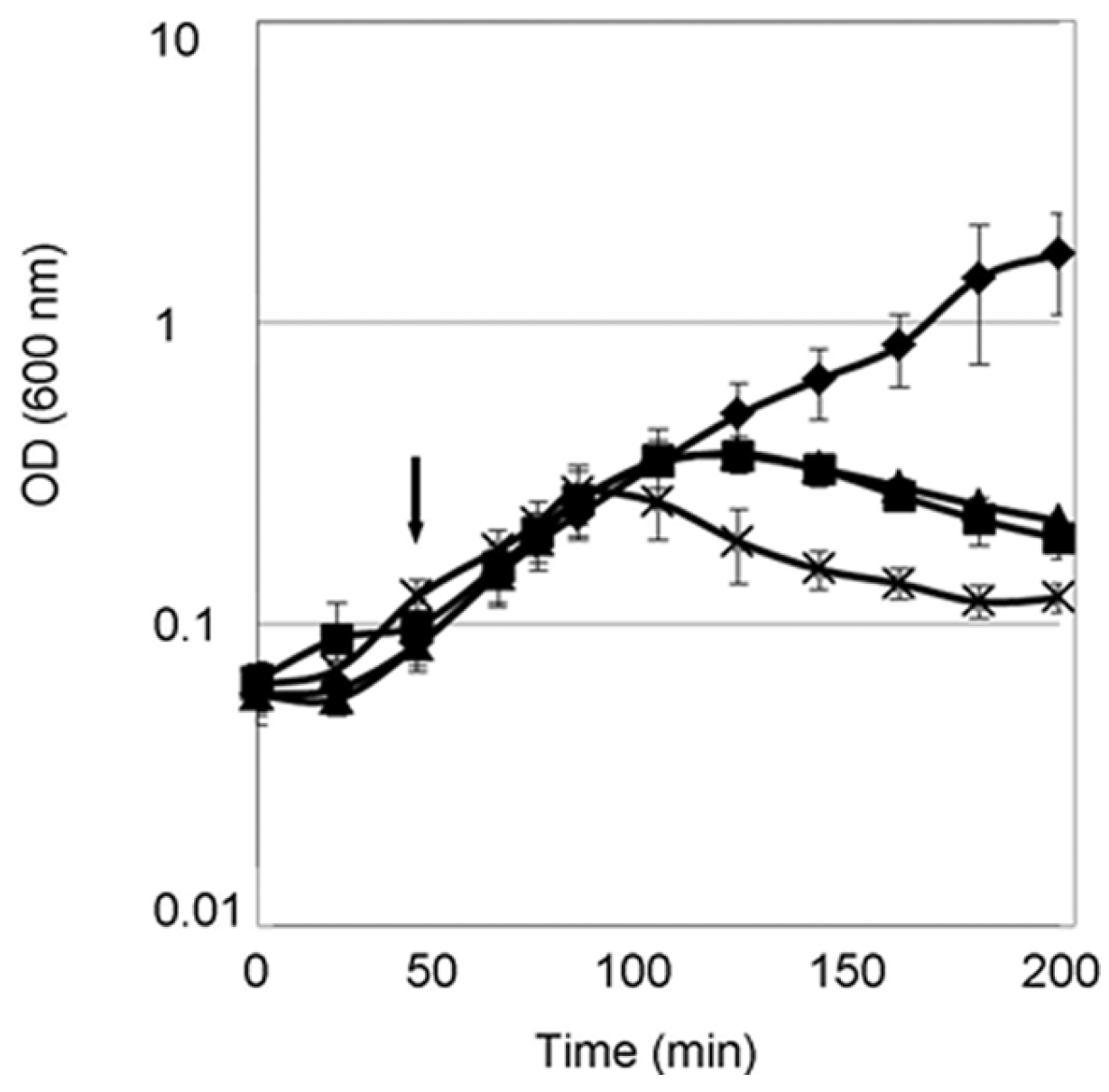
FB8, BW25113 and BW25113 ΔfkpA E. coli strains, carrying pASK-derived plasmids for periplasmic expression of full-length, truncated or mutant PcaM1, were grown aerobically at 37 °C in 2YT-ampicillin medium (50-mL cultures). When the OD600 reached 0.2, anhydrotetracycline was added at various final concentrations (from 0–400 ng/mL), and cell growth was followed by monitoring the absorbance at constant time intervals.
4.7. CFU
E. coli FB8 strains carrying the pASK plasmids allowing expression of the wild-type and D222A mutant forms of PcaM1 were grown in 2YT medium, and cultures were induced or not with 200 ng/mL of anhydrotetracycline when the OD600 reached 0.2. Samples were taken every 20 min and plated on 2YT agar after appropriate serial dilutions. Colonies were counted after overnight incubation at 37 °C. These experiments were performed in triplicate, for both strains and both induced and uninduced conditions.
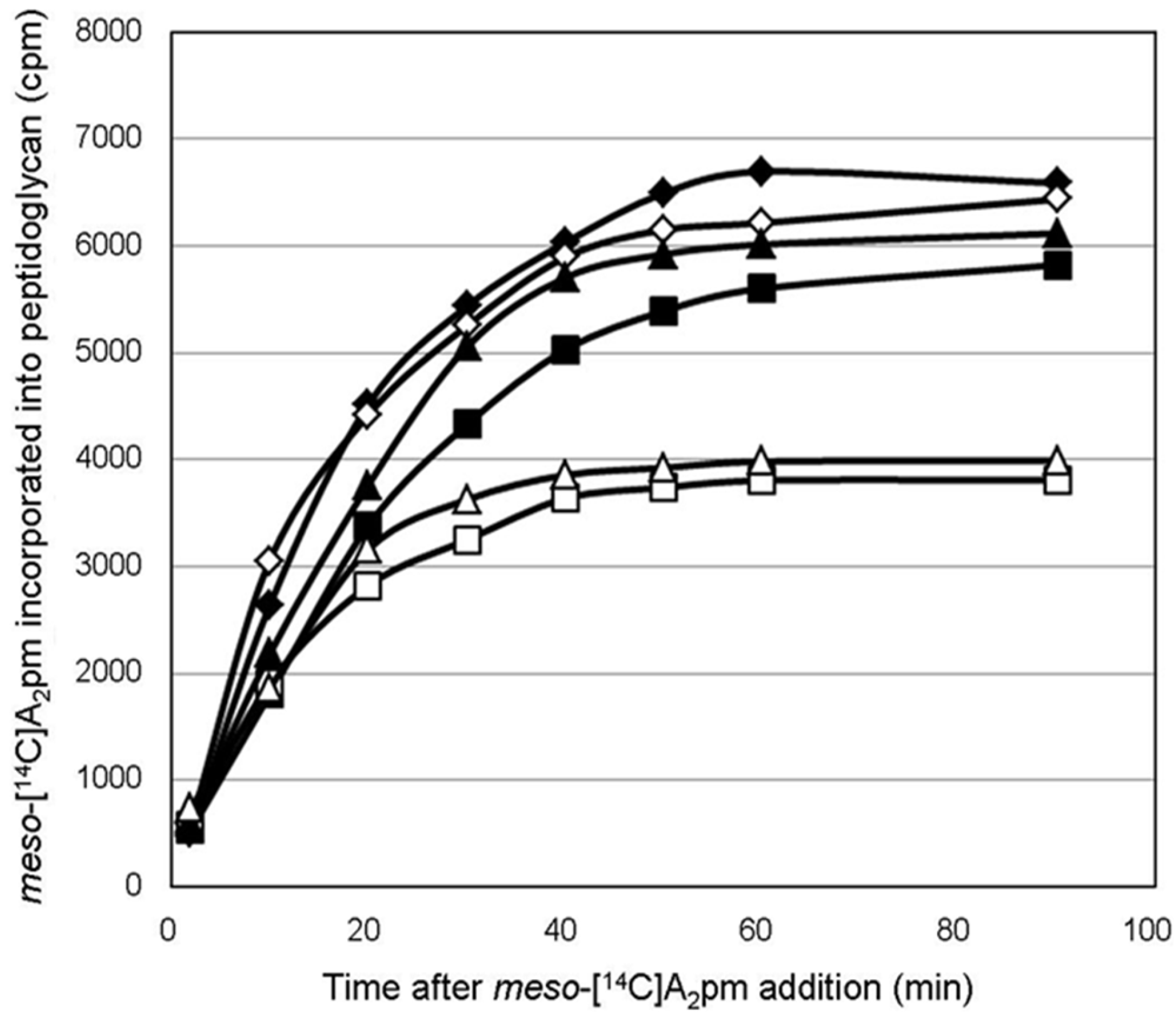
4.8. Peptidoglycan Labeling Experiments with Radioactive meso-[14C]A2pm
To determine whether cells expressing wild-type or mutant forms of PcaM1 in the periplasm were truly impaired in peptidoglycan biosynthesis, the rate of incorporation of
meso-[
14C]A
2pm into the peptidoglycan of
E. coli FB8 Δ
lysA::kan strain transformed with pASK-IBA4, pMLD381 or pMLD395 expression plasmid was followed. The latter strains were grown exponentially in minimum M63 medium (30-mL cultures) supplemented with 0.4% glucose and 100 µg/mL each of lysine, methionine and threonine [
2,
24]. In fact, only lysine is required to complement the
lysA::kan mutation of these strains, but the addition of methionine and threonine was used here to decrease as much as possible the internal cellular pool of A
2pm, as described previously [
24,
31]. When the OD
600 reached 0.2, cultures were treated with anhydrotetracycline (100 ng/mL), and
meso-[
14C]A
2pm (0.2 kBq/mL) was added 10 min later. The incorporation of
meso-[
14C]A
2pm into the peptidoglycan polymer was then followed as described previously [
2]. Briefly, 1-mL culture samples were collected regularly over time and added to 10 mL of ice-cold 5% trichloroacetic acid (TCA). Suspensions were kept on ice for 60 min, and the TCA-insoluble radiolabeled peptidoglycan material was then filtered over Whatman GF/C glass fiber filters. The filters were washed with 5% TCA and dried, and the radioactivity was counted with a liquid scintillation spectrophotometer after their immersion in a solvent system consisting of 2 mL of water and 13 mL of Unisafe 1 scintillator (Zinsser Analytic, Maidenhead, UK).
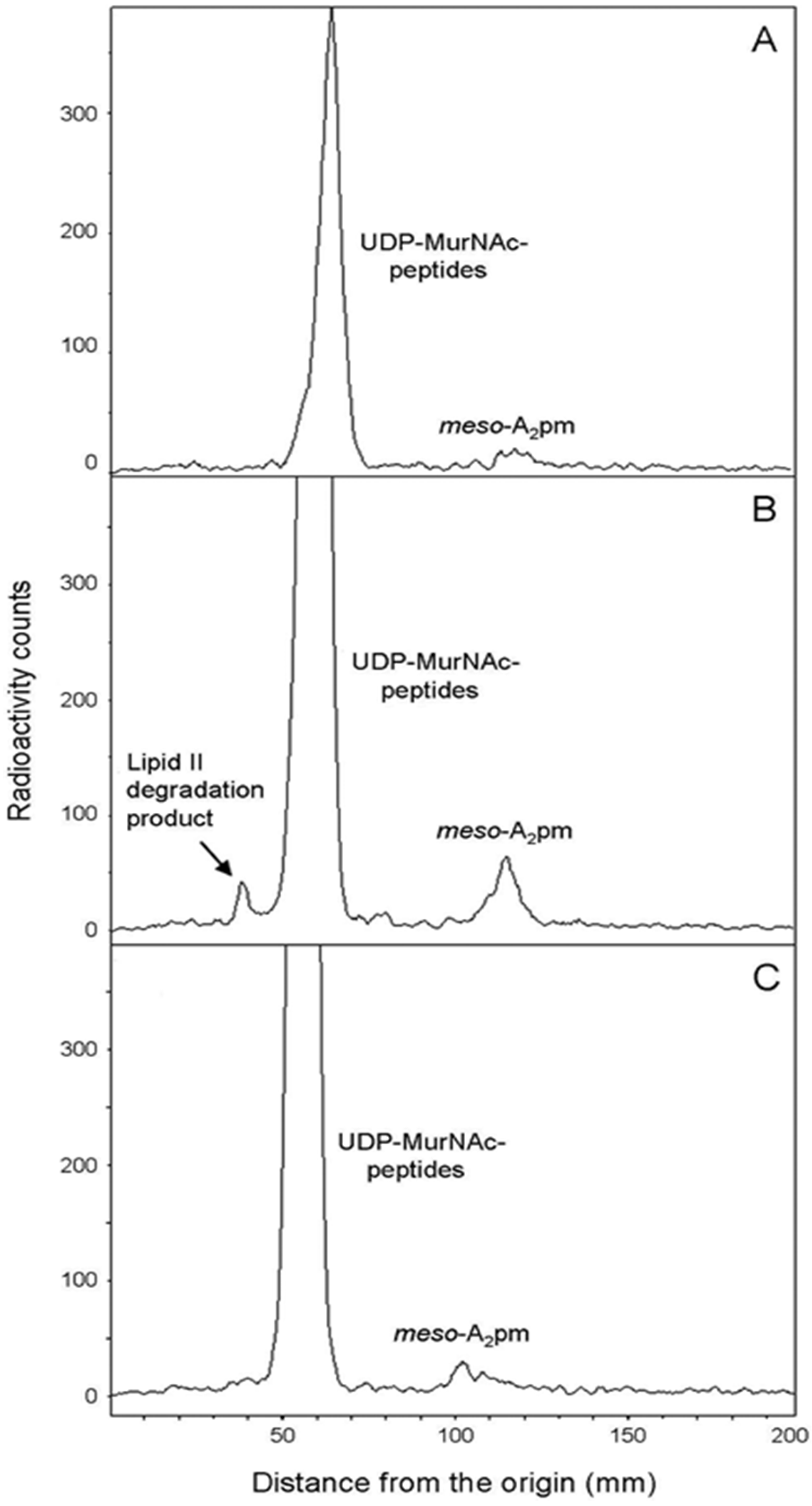
4.9. Cellular Distribution of meso-[14C]A2pm
To identify the step in peptidoglycan synthesis that was affected following periplasmic expression of the PcaM1 variants, the cellular distribution of [
14C]A
2pm incorporated in these strains was analyzed in more detail. Fifty-milliliter cultures of FB8 Δ
lysA strain transformed with pASK-IBA4, pMLD381 or pMLD395 expression plasmids were performed as above in M63-glucose minimal medium supplemented with lysine, threonine and methionine. At an OD
600 of 0.2, anhydrotetracycline (100 ng/mL) was added and
meso-[
14C]A
2pm (0.2 kBq/mL) 20 min thereafter. After 30 min of labeling, cultures were rapidly chilled to 0–4 °C, collected by centrifugation and the cell pellets suspended in 3 mL of boiling water. After 15 min at 100 °C, suspensions were chilled and centrifuged at 200,000×
g for 20 min. The supernatant was lyophilized, and both the soluble (supernatant) and insoluble (pellet) fractions were suspended in 250 µL of water. The total radioactivity recovered in these two cell fractions was measured. The insoluble fraction is known to contain peptidoglycan and lipid intermediates I and II, as well as the soluble fraction
meso-[
14C]A
2pm and the nucleotide precursors as labeled compounds [
2]. Aliquots were analyzed by TLC on silica gel plates with 1-propanol/ammonium hydroxide/water (6:3:1; v/v/v) as the mobile phase. Under such conditions, peptidoglycan remains at the origin, and UDP-MurNAc-peptides,
meso-[
14C]A
2pm and lipid intermediates migrated with
Rf values of 0.35, 0.55 and 0.7, respectively. An additional spot (
Rf of 0.3) corresponding to the lipid II degradation product 1-PP-MurNAc(pentapeptide)-GlcNAc was observed following the induction of PcaM1 expression.
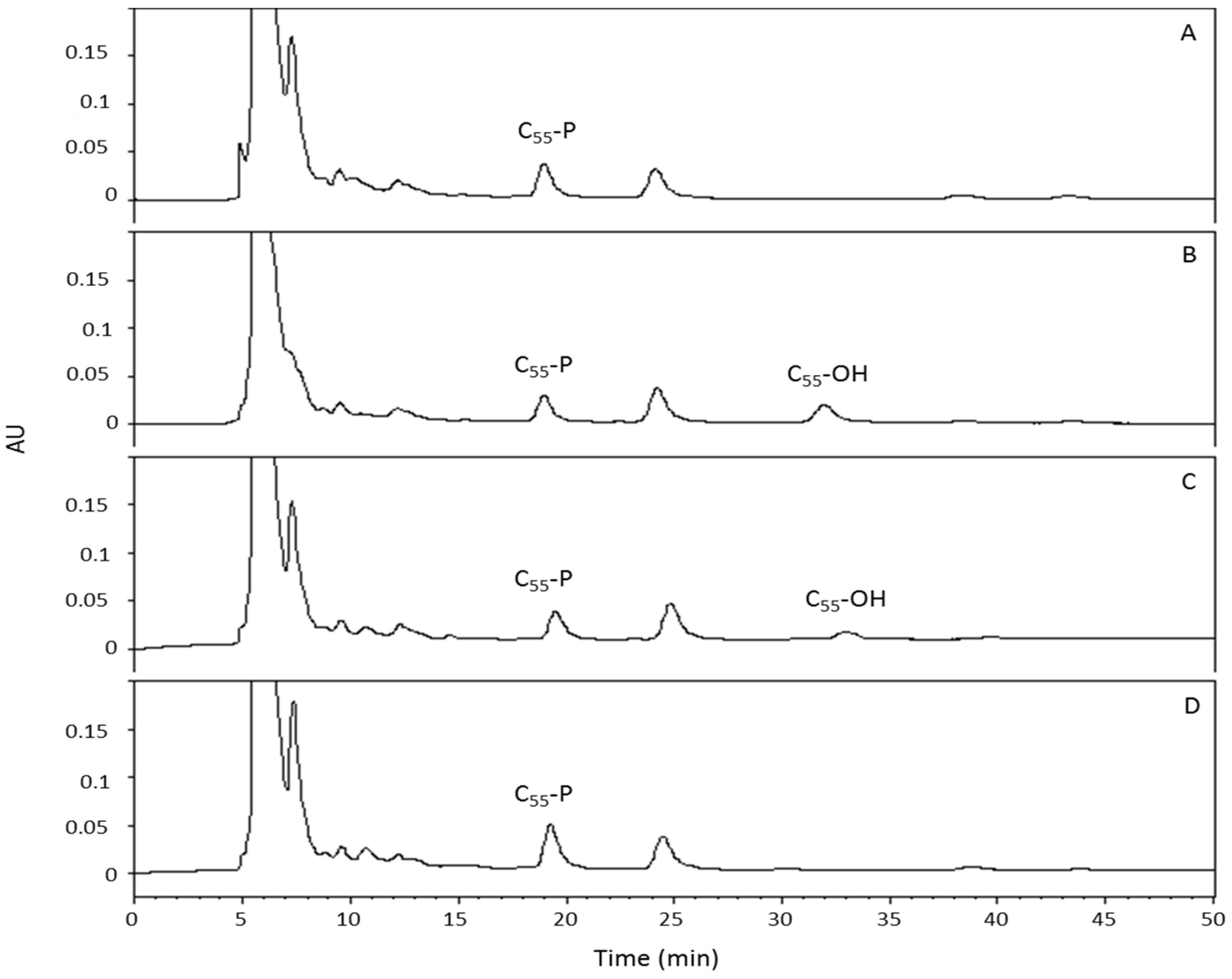
4.10. Quantitation of C55-P and Its Derivatives in Membranes of E. coli Strains Expressing Periplasmic PcaM1 and Derivatives
Cultures (100 mL) of
E. coli strain FB8 expressing the different periplasmic PcaM1 variants were grown as described above, and cells were harvested just before the onset of lysis, i.e., about 40 min after the induction of protein expression by anhydrotetracycline. Isoprenoid extraction was performed from washed membrane cell pellets of the different tested strains according to two different procedures, as previously described [
23]. Briefly, each culture was divided in two 50 mL samples, which were treated according to the Bligh and Dyer procedure [
32] to directly quantify C
55-P and C
55-OH pool levels, or to Kato’s procedure [
33], to determine the C
55-PP pool level (through this procedure, C
55-PP is totally converted into C
55-P). The resulting isoprenoid-containing organic phases were subsequently submitted to HPLC analysis for quantitation, using an isocratic elution system (2-propanol:methanol 1:4 (v/v) containing 10 mM phosphoric acid) on a reverse-phase Nucleosil C18 column (5 µm, 250 × 4.6 mm). The flow rate was 0.6 mL/min, and the quantitation of isoprenoids, monitored at 210 nm, was performed with respect to commercial compounds previously injected as standards in the same conditions.
4.11. Optical Microscopy Analyses
Bacteria were visualized using a DMIRE2 optical microscope (Leica) equipped with a CCD camera (CoolSNAP HQ2, Roper Scientific, Martinsried, Germany).
4.12. MALDI-TOF Mass Spectrometry
MALDI-TOF mass spectra of the wild-type PcaM1 were recorded in the linear mode with delayed extraction on a PerSeptive Voyager-DE STR instrument (Applied Biosystems, Carlsbad, CA, USA) equipped with a 337-nm laser. Buffer and glycerol were removed from the samples by using a ZipTip C4 pipette tip (Merck Millipore, Molsheim, France) according to the manufacturer’s recommendations with slight modifications. Briefly, the bacteriocin was adsorbed on ZipTip, and after it was washed with 0.1% trifluoroacetic acid (TFA), the bacteriocin was eluted with 7.5 µL of 0.1% TFA in 70% acetonitrile. Subsequently, 1 µL of matrix solution (10 mg/mL sinapinic acid in 0.1% TFA-acetonitrile (70:30, v/v)) was deposited on the plate, followed by 0.3, 0.5 or 1 µL of concentrated bacteriocin. After evaporation of the solvents, spectra were recorded in the positive mode at an acceleration voltage of +25 kV and an extraction delay time of 300 ns. Carbonic anhydrase was used as an external calibrant.
4.13. Chemicals
[
14C]lipid II labeled in the GlcNAc moiety was prepared as described previously [
2]. The lipid II used in this study was C
55-PP-MurNAc(
l-Ala-γ-
d-Glu-
meso-A
2pm-
d-Ala-
d-Ala)-GlcNAc, where
meso-A
2pm represents
meso-diaminopimelic acid. C
55-PP, C
55-P and C
55-OH were purchased from the Institute of Biochemistry and Biophysics of the Polish Academy of Sciences (Warsaw, Poland) and
meso-[
14C]A
2pm from the Commissariat à l’Energie Atomique (Saclay, France).
N-Dodecyl-
β-
d-maltopyranoside (DDM) was from Anatrace (Maumee, OH, USA), isopropyl-
β-
d-thiogalactopyranoside (IPTG) from Eurogentec (Angers, France) and Ni
2+-nitrilotriacetate agarose from Qiagen (Courtaboeuf, France). Antibiotics and reagents were from Sigma-Aldrich (Saint-Quentin Fallavier, France). Synthesis of oligonucleotides and DNA sequencing were done by Eurofins Genomics (Ebersberg, Germany).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}