Fragment-Based Discovery of Inhibitors of the Bacterial DnaG-SSB Interaction


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Screening of Fragment Libraries
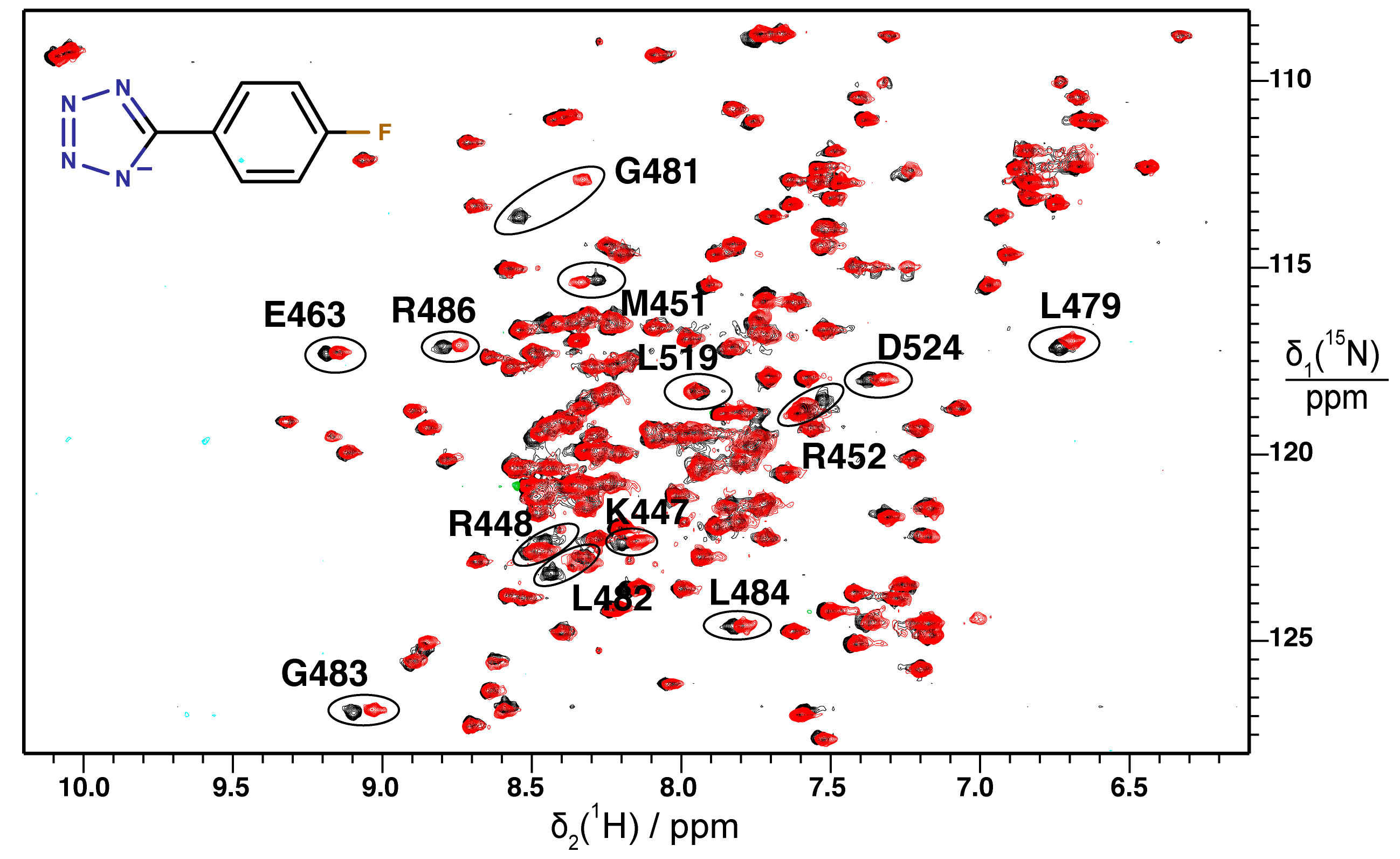
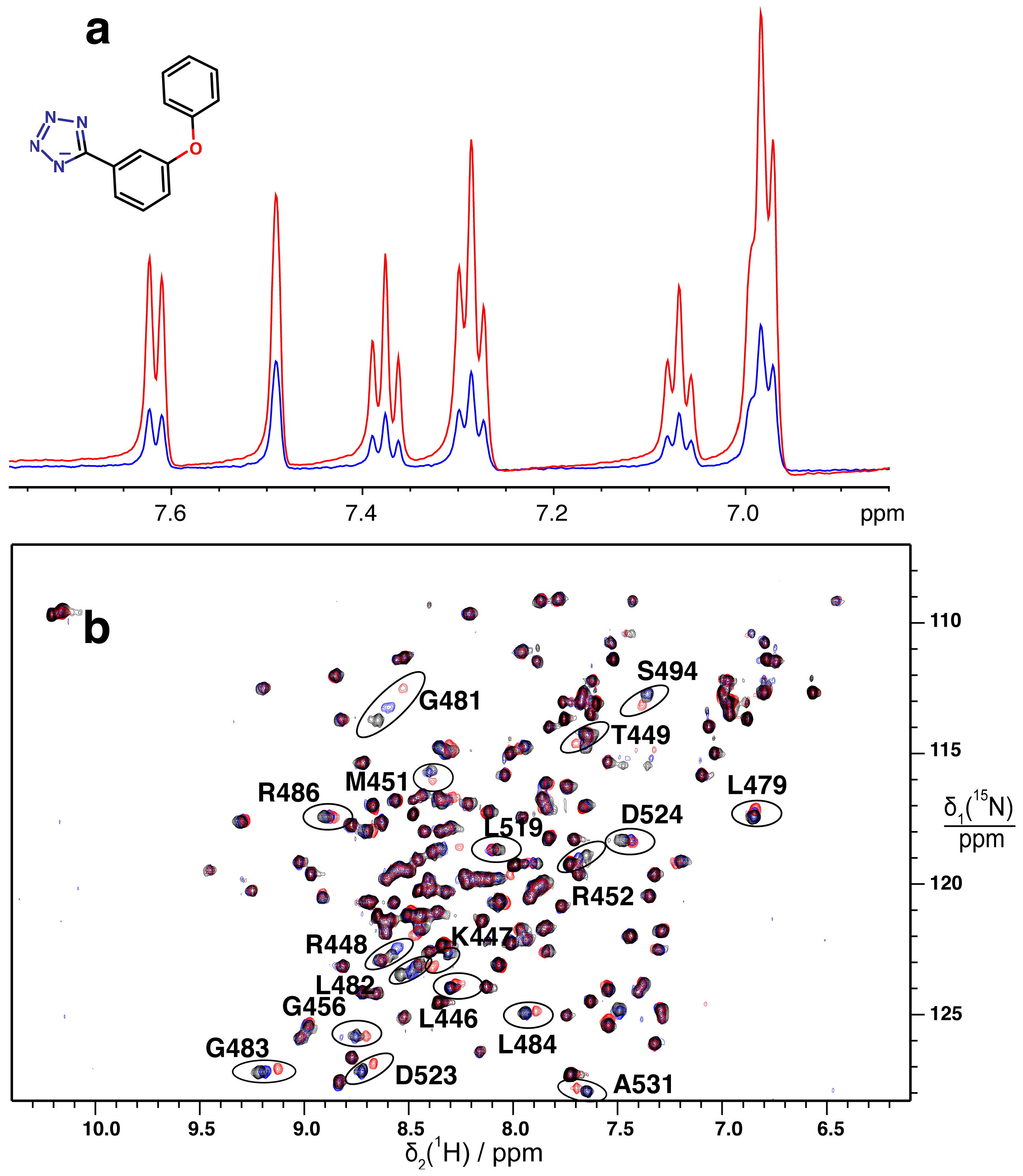
2.2. Validation of Fragment Binding by 2D NMR
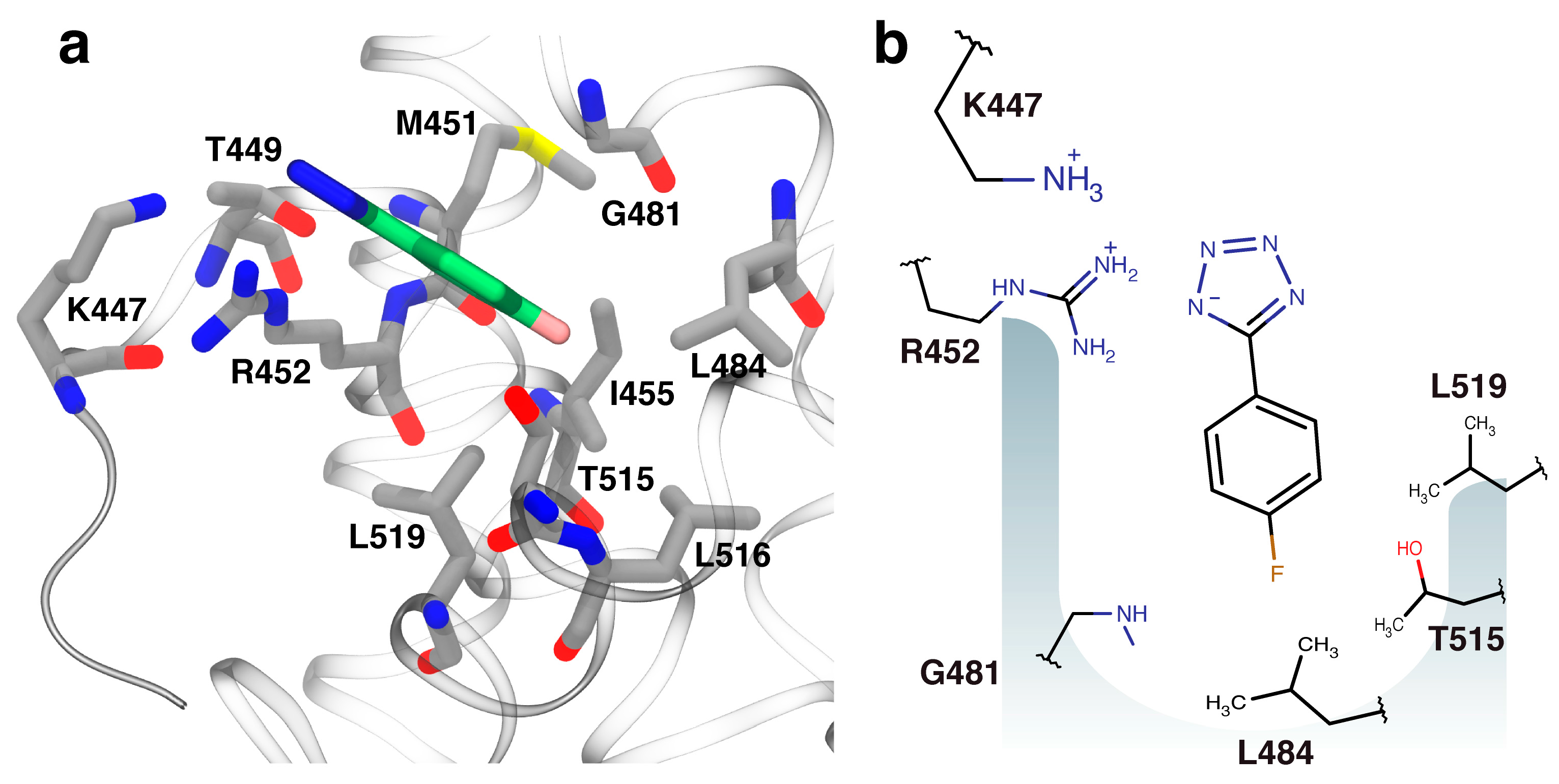
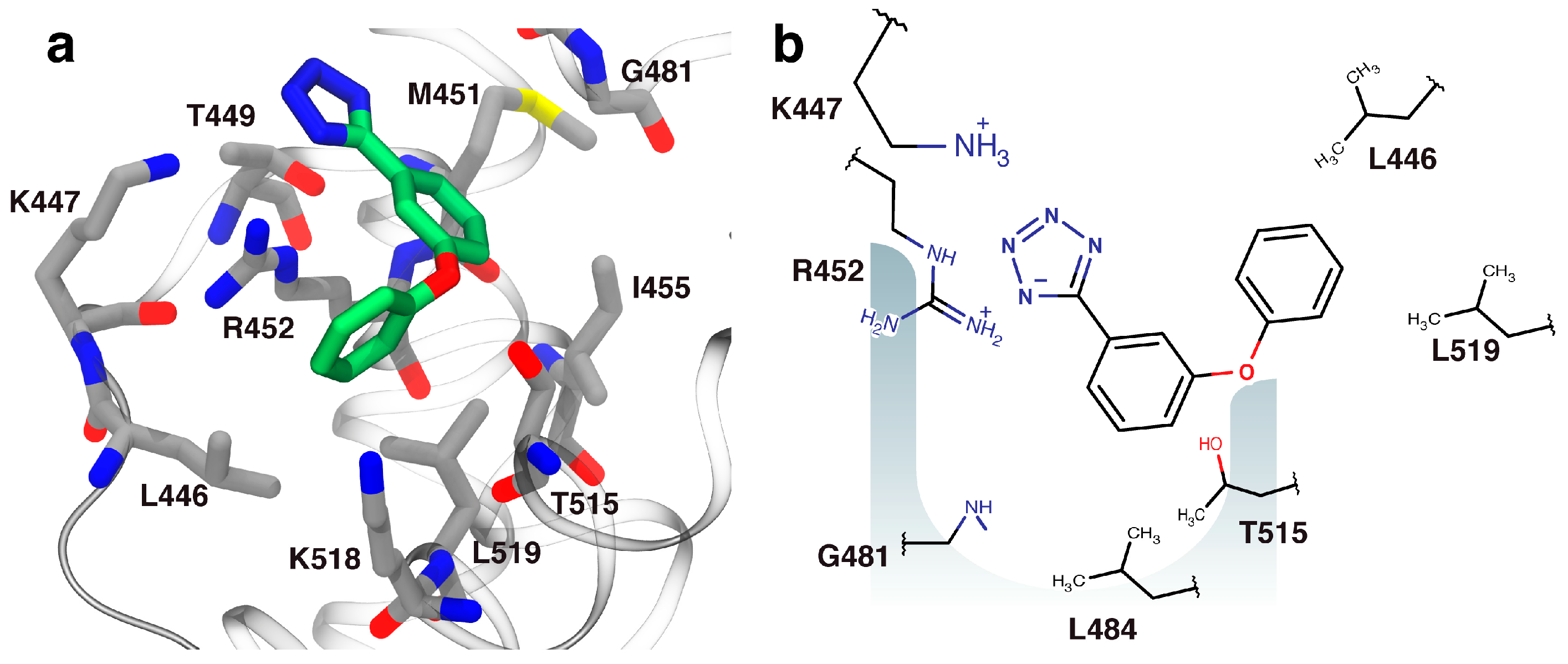
2.3. Orientation of Identified Hits Using Molecular Docking
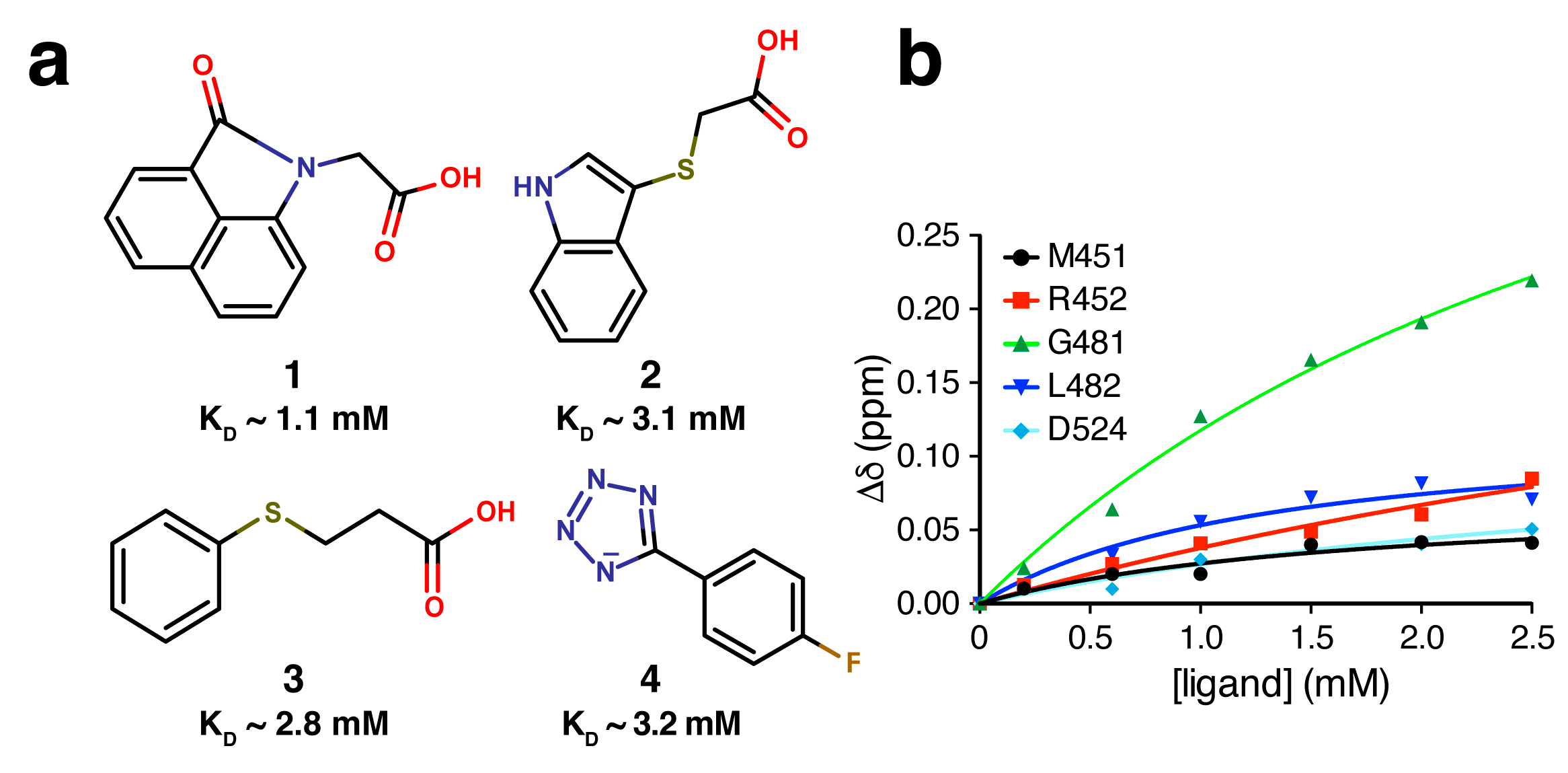
2.4. Fragment-to-Hit Optimization
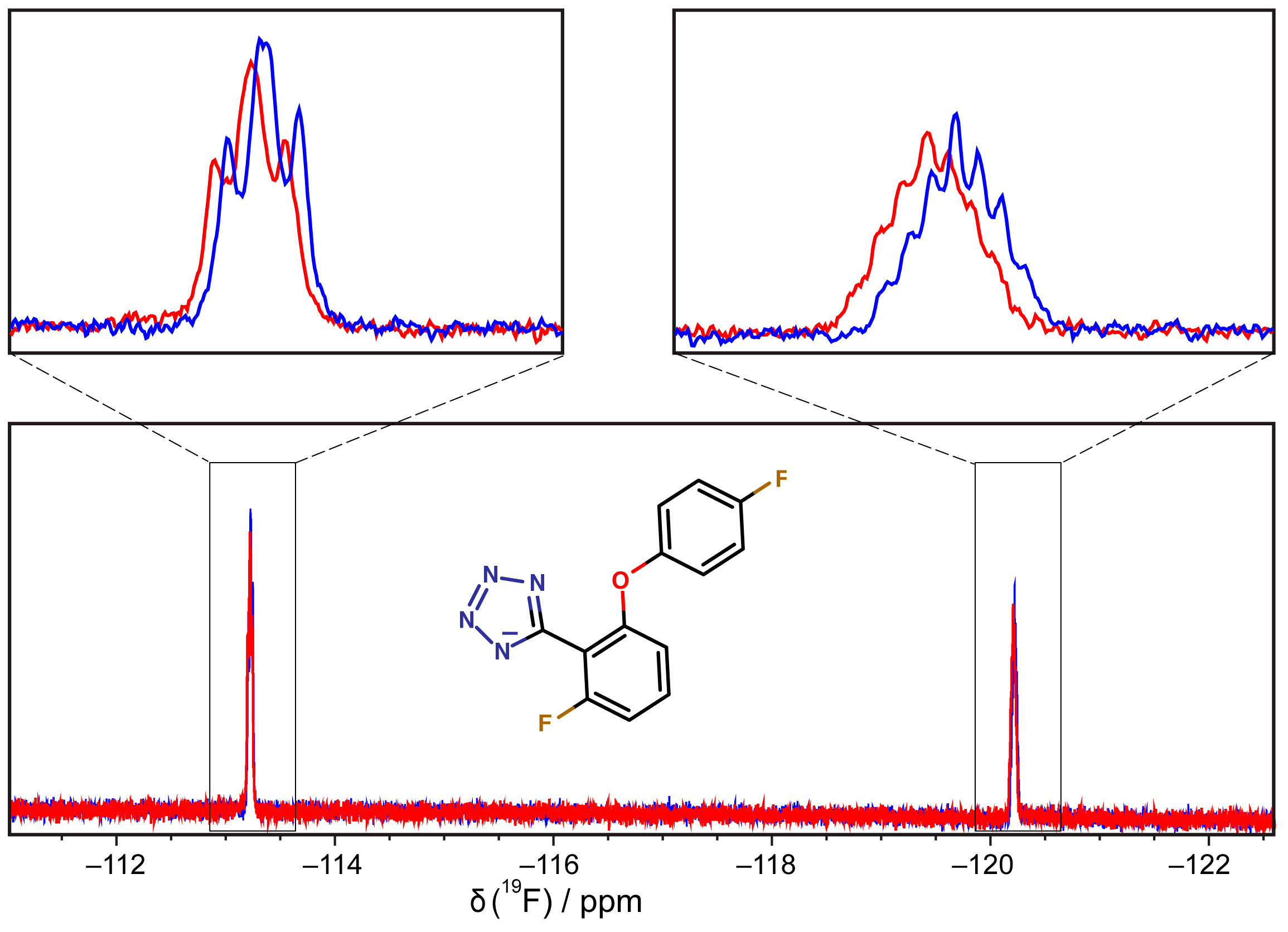
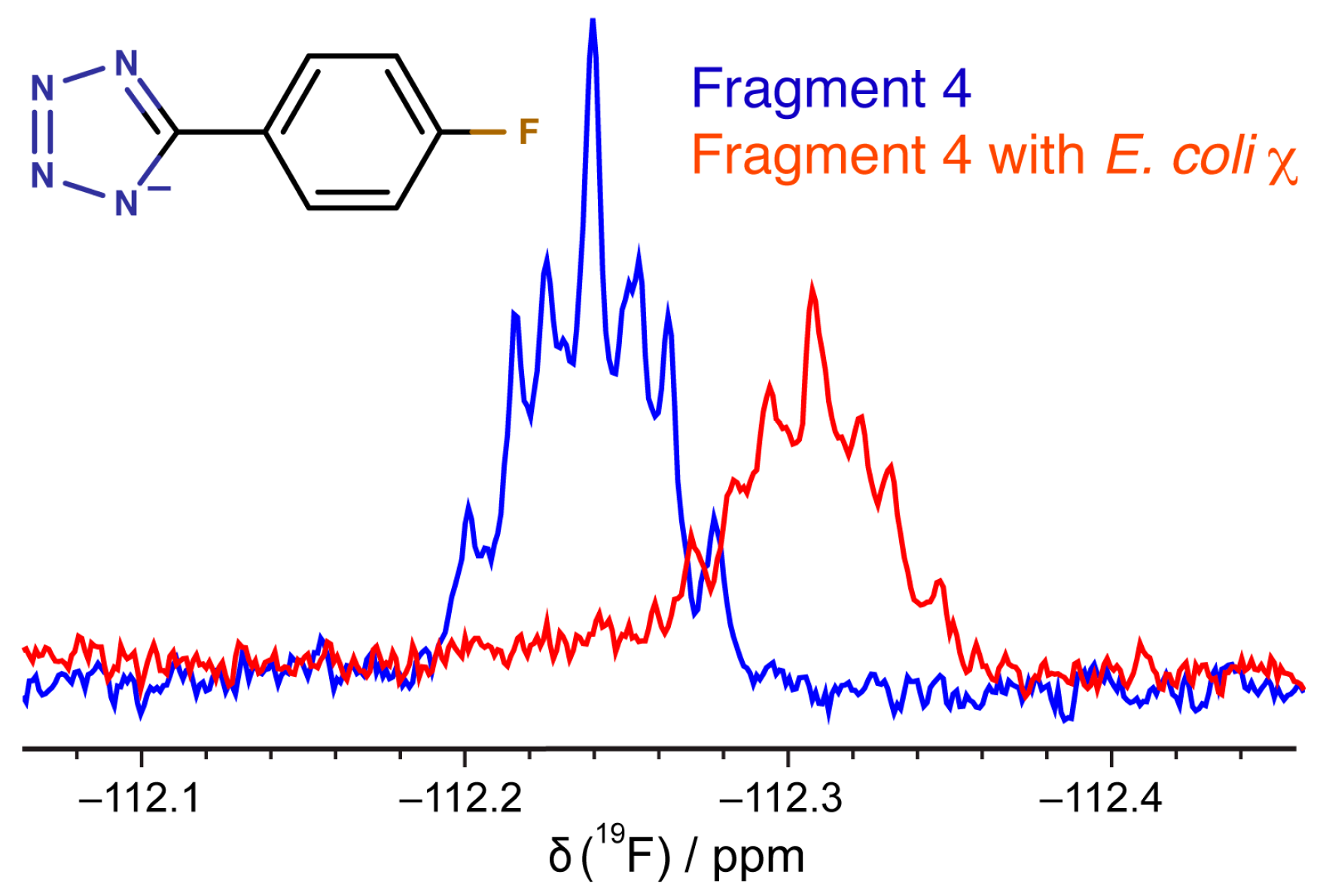
2.5. Binding of Compounds to Other SSB Partner Proteins
3. Materials and Methods
3.1. Protein Expression and Purification
3.2. Fragment Libraries
3.3. SPR Competition Assay
3.4. Saturation-Transfer Difference (STD) NMR Spectroscopy
3.5. 2D 15N–1H HSQC Spectra
3.6. 19F-NMR Spectroscopy
3.7. Molecular Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kobori, J.A.; Kornberg, A. The Escherichia coli dnaC gene product. II. Purification, physical properties, and role in replication. J. Biol. Chem. 1982, 257, 13763–13769. [Google Scholar] [PubMed]
- Kobori, J.A.; Kornberg, A. The Escherichia coli dnaC gene product. III. Properties of the dnaB–dnaC protein complex. J. Biol. Chem. 1982, 257, 13770–13775. [Google Scholar] [PubMed]
- Tougu, K.; Marians, K.J. The interaction between helicase and primase sets the replication fork clock. J. Biol. Chem. 1996, 271, 21398–21405. [Google Scholar] [CrossRef] [PubMed]
- Kitani, T.; Yoda, K.; Ogawa, T.; Okazaki, T. Evidence that discontinuous DNA replication in Escherichia coli is primed by approximately 10 to 12 residues of RNA starting with a purine. J. Mol. Biol. 1985, 184, 45–52. [Google Scholar] [CrossRef]
- Yoda, K.-Y.; Yasuda, H.; Jiang, X.-W.; Okazaki, T. RNA-primed intitiation sites of DNA replication in the origin region of bacteriophage λ genome. Nucleic Acids Res. 1998, 16, 6531–6546. [Google Scholar] [CrossRef]
- Bouché, J.P.; Zechel, K.; Kornberg, A. dnaG gene product, a rifampicin-resistant RNA polymerase, initiates the conversion of a single-stranded coliphage DNA to its duplex replicative form. J. Biol. Chem. 1975, 250, 5995–6001. [Google Scholar] [PubMed]
- Pan, H.; Wigley, D.B. Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Structure 2000, 8, 231–239. [Google Scholar] [CrossRef]
- Keck, J.L.; Berger, J.M. Structure of the RNA polymerase domain of E. coli primase. Science 2000, 287, 2482–2486. [Google Scholar] [CrossRef] [PubMed]
- Podobnik, M.; McInerney, P.; O’Donnell, M.; Kuriyan, J. A TOPRIM domain in the crystal structure of the catalytic core of Escherichia coli primase confirms a structural link to DNA topoisomerases. J. Mol. Biol. 2000, 300, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Godson, G.N. Interaction of Escherichia coli primase with a phage G4oric-E. coli SSB complex. J. Bacteriol. 1996, 178, 6701–6705. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tougu, K.; Marians, K.J. The extreme C terminus of primase is required for interaction with DnaB at the replication fork. J. Biol. Chem. 1996, 271, 21391–21397. [Google Scholar] [CrossRef] [PubMed]
- Yuzhakov, A.; Kelman, Z.; O’Donnell, M. Trading places on DNA–a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 1999, 96, 153–163. [Google Scholar] [CrossRef]
- Oakley, A.J.; Loscha, K.V.; Schaeffer, P.M.; Liepinsh, E.; Pintacuda, G.; Wilce, M.C.J.; Otting, G.; Dixon, N.E. Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J. Biol. Chem. 2005, 280, 11495–11504. [Google Scholar] [CrossRef] [PubMed]
- Su, X.-C.; Schaeffer, P.M.; Loscha, K.V.; Gan, P.H.P.; Dixon, N.E.; Otting, G. Monomeric solution structure of the helicase-binding domain of Escherichia coli DnaG primase. FEBS J. 2006, 273, 4997–5009. [Google Scholar] [CrossRef] [PubMed]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Causer, R.J.; Dixon, N.E. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets 2012, 13, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Glover, B.P.; McHenry, C.S. The χψ subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem. 1998, 273, 23476–23484. [Google Scholar] [CrossRef] [PubMed]
- Kelman, Z.; Yuzhakov, A.; Andjelkovic, J.; O’Donnell, M. Devoted to the lagging strand–the χ subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO J. 1998, 17, 2436–2449. [Google Scholar] [CrossRef] [PubMed]
- Witte, G. DNA polymerase III χ subunit ties single-stranded DNA binding protein to the bacterial replication machinery. Nucleic Acids Res. 2003, 31, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, R.; Witte, G.; Urbanke, C.; Manstein, D.J.; Curth, U. 3D structure of Thermus aquaticus single-stranded DNA-binding protein gives insight into the functioning of SSB proteins. Nucleic Acids Res. 2006, 34, 6708–6717. [Google Scholar] [CrossRef] [PubMed]
- Naue, N.; Fedorov, R.; Pich, A.; Manstein, D.J.; Curth, U. Site-directed mutagenesis of the χ subunit of DNA polymerase III and single-stranded DNA-binding protein of E. coli reveals key residues for their interaction. Nucleic Acids Res. 2011, 39, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Bahng, S.; Massoni, S.C.; George, N.P.; Sandler, S.J.; Marians, K.J.; Keck, J.L. Structure of the SSB–DNA polymerase III interface and its role in DNA replication. EMBO J. 2011, 30, 4236–4247. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, B.; George, N.P.; Thurmes, T.M.; Zhou, R.; Jani, N.; Wessel, S.R.; Sandler, S.J.; Ha, T.; Keck, J.L. Structural mechanisms of PriA-mediated DNA replication restart. Proc. Natl. Acad. Sci. USA 2014, 111, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Keck, J.L. Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc. Natl. Acad. Sci. USA 2008, 105, 9169–9174. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Windsor, M.A.; Gellman, S.H.; Keck, J.L. Peptide inhibitors identify roles for SSB C-terminal residues in SSB/exonuclease I complex formation. Biochemistry 2009, 48, 6764–6771. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Naue, N.; Beerbaum, M.; Bogutzki, A.; Schmieder, P.; Curth, U. The helicase-binding domain of Escherichia coli DnaG primase interacts with the highly conserved C-terminal region of single-stranded DNA-binding protein. Nucleic Acids Res. 2013, 41, 4507–4517. [Google Scholar] [CrossRef] [PubMed]
- Ryzhikov, M.; Koroleva, O.; Postnov, D.; Tran, A.; Korolev, S. Mechanism of RecO recruitment to DNA by single-stranded DNA binding protein. Nucleic Acids Res. 2011, 39, 6305–6314. [Google Scholar] [CrossRef] [PubMed]
- Chilingaryan, Z.; Yin, Z.; Oakley, A.J. Fragment-based screening by protein crystallography: Successes and pitfalls. Int. J. Mol. Sci. 2012, 13, 12857–12879. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Doak, B.C.; Morton, C.J.; Simpson, J.S.; Scanlon, M.J. Design and evaluation of the performance of an NMR screening fragment library. Aust. J. Chem. 2013, 66, 1465–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Malik, M.A.; Wani, M.Y.; Al-Thabaiti, S.A.; Shiekh, R.A. Tetrazoles as carboxylic acid isosteres: Chemistry and biology. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 15–37. [Google Scholar] [CrossRef]
- Allen, F.H.; Groom, C.R.; Liebeschuetz, J.W.; Bardwell, D.A.; Olsson, T.S.G.; Wood, P.A. The hydrogen bond environments of 1H-tetrazole and tetrazolate rings: The structural basis for tetrazole–carboxylic acid bioisosterism. J. Chem. Inf. Model. 2012, 52, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Fielding, L. NMR methods for the determination of protein–ligand dissociation constants. Curr. Top. Med. Chem. 2007, 3, 39–53. [Google Scholar] [CrossRef]
- Lian, L.-Y. NMR studies of weak protein–protein interactions. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 71, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Vulpetti, A. Intermolecular and intramolecular hydrogen bonds involving fluorine atoms: Implications for recognition, selectivity, and chemical properties. ChemMedChem 2012, 7, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Brameld, K.A.; Kuhn, B.; Reuter, D.C.; Stahl, M. Small molecule conformational preferences derived from crystal structure data. A medicinal chemistry focused analysis. J. Chem. Inf. Model. 2008, 48, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC−A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Medek, A.; Hajduk, P.J.; Mack, J.; Fesik, S.W. The use of differential chemical shifts for determining the binding site location and orientation of protein-bound ligands. J. Am. Chem. Soc. 2000, 122, 1241–1242. [Google Scholar] [CrossRef]
- Stamford, N.P.J.; Lilley, P.E.; Dixon, N.E. Enriched sources of Escherichia coli replication proteins. The dnaG primase is a zinc metalloprotein. Biochim. Biophys. Acta 1992, 1132, 17–25. [Google Scholar] [CrossRef]
- Love, C.A.; Lilley, P.E.; Dixon, N.E. Stable high-copy-number bacteriophage λ promoter vectors for overproduction of proteins in Escherichia coli. Gene 1996, 176, 49–53. [Google Scholar] [CrossRef]
- Loscha, K.; Oakley, A.J.; Bancia, B.; Schaeffer, P.M.; Prosselkov, P.; Otting, G.; Wilce, M.C.J.; Dixon, N.E. Expression, purification, crystallization, and NMR studies of the helicase interaction domain of Escherichia coli DnaG primase. Protein Expr. Purif. 2004, 33, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Ziarek, J.J.; Peterson, F.C.; Lytle, B.L.; Volkman, B.F. Binding site identification and structure determination of protein–ligand complexes by NMR: A semiautomated approach. Methods Enzymol. 2011, 493, 241–275. [Google Scholar] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.T.Y.; Oakley, A.J.; Dixon, N.E. The DnaG primase interaction with single-stranded DNA-binding protein (SSB). Manuscript in preparation.
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chilingaryan, Z.; Headey, S.J.; Lo, A.T.Y.; Xu, Z.-Q.; Otting, G.; Dixon, N.E.; Scanlon, M.J.; Oakley, A.J. Fragment-Based Discovery of Inhibitors of the Bacterial DnaG-SSB Interaction. Antibiotics 2018, 7, 14. https://doi.org/10.3390/antibiotics7010014
Chilingaryan Z, Headey SJ, Lo ATY, Xu Z-Q, Otting G, Dixon NE, Scanlon MJ, Oakley AJ. Fragment-Based Discovery of Inhibitors of the Bacterial DnaG-SSB Interaction. Antibiotics. 2018; 7(1):14. https://doi.org/10.3390/antibiotics7010014
Chicago/Turabian StyleChilingaryan, Zorik, Stephen J. Headey, Allen T. Y. Lo, Zhi-Qiang Xu, Gottfried Otting, Nicholas E. Dixon, Martin J. Scanlon, and Aaron J. Oakley. 2018. "Fragment-Based Discovery of Inhibitors of the Bacterial DnaG-SSB Interaction" Antibiotics 7, no. 1: 14. https://doi.org/10.3390/antibiotics7010014
APA StyleChilingaryan, Z., Headey, S. J., Lo, A. T. Y., Xu, Z.-Q., Otting, G., Dixon, N. E., Scanlon, M. J., & Oakley, A. J. (2018). Fragment-Based Discovery of Inhibitors of the Bacterial DnaG-SSB Interaction. Antibiotics, 7(1), 14. https://doi.org/10.3390/antibiotics7010014