Complementation Studies of Bacteriophage ? O Amber Mutants by Allelic Forms of O Expressed from Plasmid, and O-P Interaction Phenotypes


Abstract
:1. Introduction
2. Results
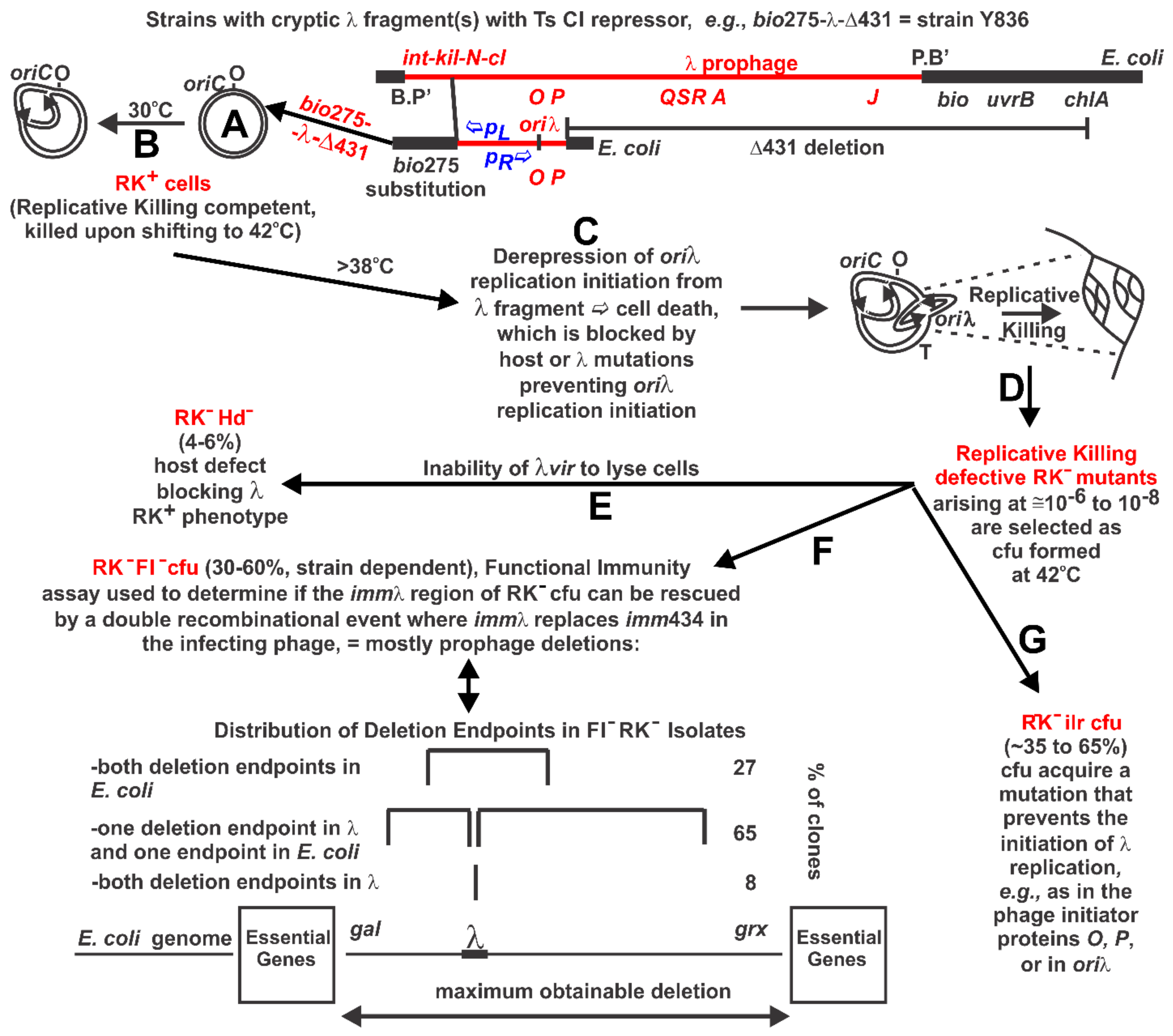
2.1. Taking Stock of O Mutations in Phage and Prophage Collections
2.2. Replicative Killing Selection and Mutants
2.3. Complementation for O Activity in Trans
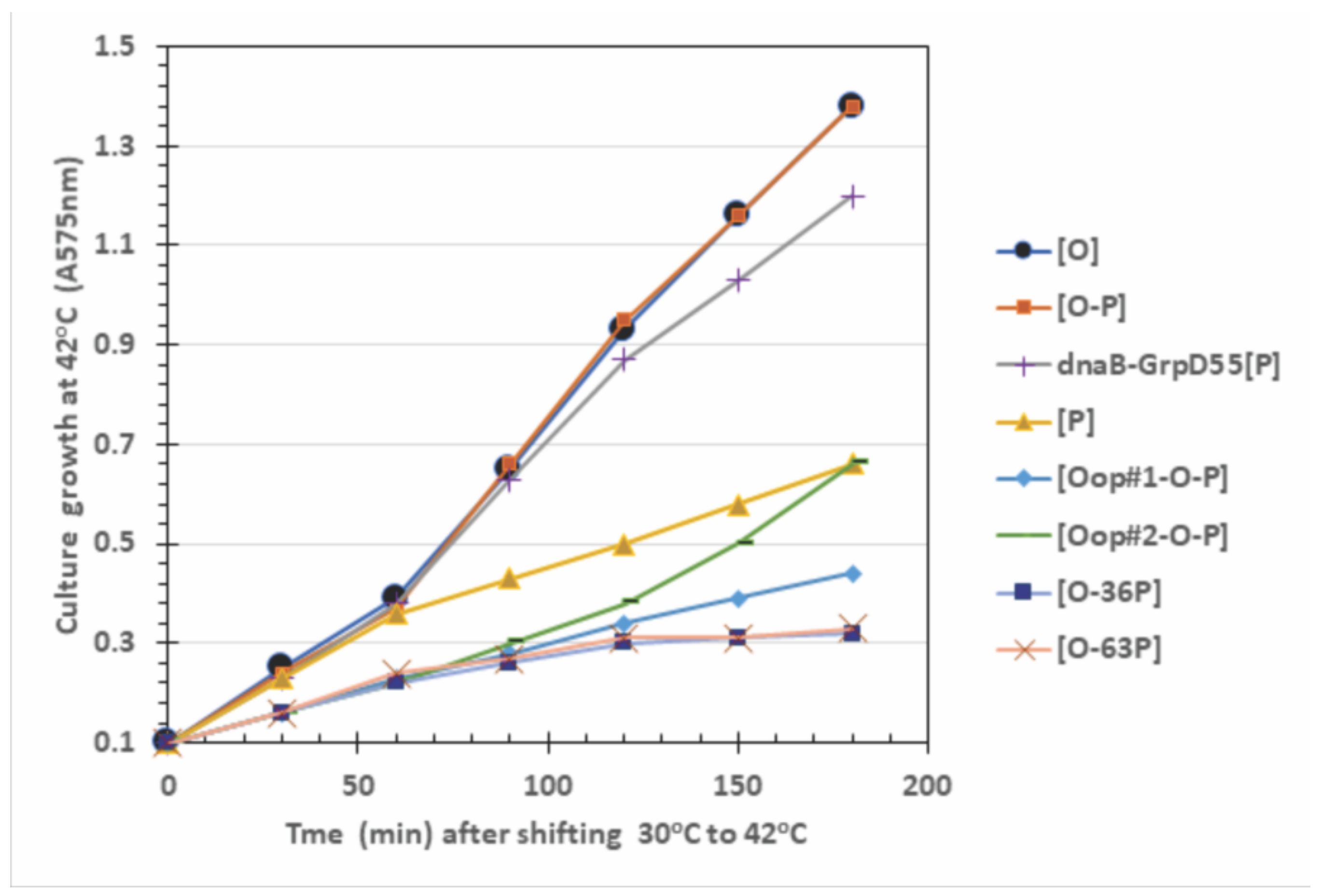
2.4. Influence of O-P Co-Expression on Cell Growth, P-Lethality, and Plasmid Loss
3. Discussion
3.1. O-Complementation
3.2. O:P Interaction Effects
3.3. RK− Mutant Selection Considerations
4. Materials and Methods
4.1. Complementation Assays and Initial Strategy for Characterizing RK− Mutants
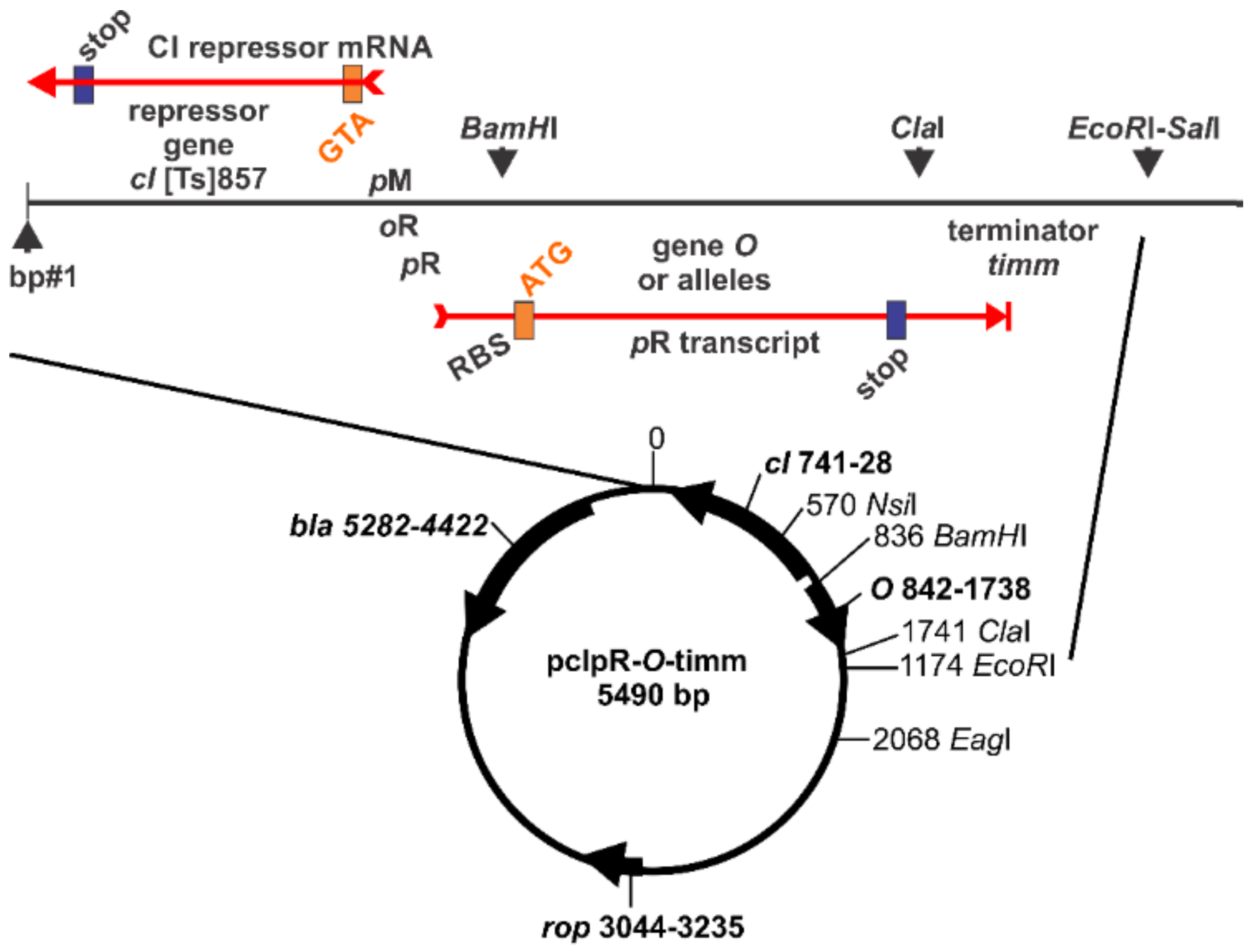
4.2. DNA Sequence Analysis of λ Phage, Prophage and Plasmid Constructs
4.3. Plasmid Constructs
4.4. Bacterial and Phage Strains
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Casjens, S.R.; Hendrix, R.W. Bacteriophage lambda: Early pioneer and still relevant. Virology 2015, 479–480, 310–330. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.I.; Gottesman, M. Lytic mode of lambda development. In Lambda II; Hendrix, R.W., Roberts, J.W., Stahl, F.W., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1983; pp. 21–51. [Google Scholar]
- Hendrix, R.W.; Casjens, S. Bacteriophage lambda and its genetic neighborhood. In The Bacteriophages, 2nd ed.; Calendar, R., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 409–447. [Google Scholar]
- Court, D.L.; Oppenheim, A.B.; Adhya, S.L. A new look at bacteriophage lambda genetic networks. J. Bacteriol. 2007, 189, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Tsurimoto, T.; Matsubara, K. Purified bacteriophage lambda O protein binds to four repeating sequences at the lambda replication origin. Nucleic Acids Res. 1981, 9, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Tsurimoto, T.; Matsubara, K. Purification of bacteriophage lambda O protein that specifically binds to the origin of replication. Mol. Gen. Genet. 1981, 181, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.B.; Biswas, E.E. Regulation of dnaB function in DNA replication in Escherichia coli by dnaC and lambda P gene products. J. Biol. Chem. 1987, 262, 7831–7838. [Google Scholar] [PubMed]
- Alfano, C.; McMacken, R. Ordered assembly of nucleoprotein structures at the bacteriophage lambda replication origin during the initiation of DNA replication. J. Biol. Chem. 1989, 264, 10699–10708. [Google Scholar] [PubMed]
- Zylicz, M.; Ang, D.; Liberek, K.; Georgopoulos, C. Initiation of lambda DNA replication with purified host- and bacteriophage-encoded proteins: The role of the dnaK, dnaJ and grpE heat shock proteins. EMBO J. 1989, 8, 1601–1608. [Google Scholar] [PubMed]
- Thomas, R.; Bertani, L.E. On the Control of the Replication of Temperate Bacteriophages Superinfecting Immune Hosts. Virology 1964, 24, 241–253. [Google Scholar] [CrossRef]
- Hayes, S.; Hayes, C. Spontaneous lambda OR mutations suppress inhibition of bacteriophage growth by nonimmune exclusion phenotype of defective lambda prophage. J. Virol. 1986, 58, 835–842. [Google Scholar] [PubMed]
- Furth, M.E.; Dove, W.F.; Meyer, B.J. Specificity determinants for bacteriophage lambda DNA replication. III. Activation of replication in lambda ric mutants by transcription outside of ori. J. Mol. Biol. 1982, 154, 65–83. [Google Scholar] [CrossRef]
- Moore, D.D.; Blattner, F.R. Sequence of lambda ric5b. J. Mol. Biol. 1982, 154, 81–83. [Google Scholar] [CrossRef]
- Echols, H.; Guarneros, G. Control of Integration and Excision. In Lambda II; Hendrix, R.W., Roberts, J.W., Stahl, F.W., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1983; pp. 75–92. [Google Scholar]
- Pereira da Silva, L.; Eisen, H.; Jacob, F. Sur la replication du bacteriophage. C. R. Acad. Sci. Paris 1968, 266, 926–928. [Google Scholar]
- Brachet, P.; Eisen, H.; Rambach, A. Mutations of coliphage lambda affecting the expression of replicative functions O and P. Mol. Gen. Genet. 1970, 108, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Castellazzi, M.; Brachet, P.; Eisen, H. Isolation and characterization of deletions in bacteriophage lambda residing as prophage in E. coli K 12. Mol. Gen. Genet. 1972, 117, 211–218. [Google Scholar] [PubMed]
- Dove, W.F.; Inokuchi, H.; Stevens, W.F. Replication control in phage lambda. In The Bacteriophage Lambda; Hershey, A.D., Ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1971; pp. 747–771. [Google Scholar]
- Lieb, M. Studies of heat-inducible lambda-phage. 3. Mutations in cistron N affecting heat induction. Genetics 1966, 54, 835–844. [Google Scholar] [PubMed]
- Rambach, A. Replicator mutants of bacteriophage lambda: Characterization of two subclasses. Virology 1973, 54, 270–277. [Google Scholar] [CrossRef]
- Sly, W.S.; Eisen, H.A.; Siminovitch, L. Host survival following infection with or induction of bacteriophage lambda mutants. Virology 1968, 34, 112–127. [Google Scholar] [CrossRef]
- Hayes, S. Mutations suppressing loss of replication control: Genetic analysis of bacteriophage lambda-dependent replicative killing, replication initiation, and mechanisms of mutagenesis. In DNA Replication and Mutagenesis; Moses, R.E., Summers, W.C., Eds.; American Society for Microbiology: Washington, DC, USA, 1988; pp. 367–377. [Google Scholar]
- Greer, H. The kil gene of bacteriophage lambda. Virology 1975, 66, 589–604. [Google Scholar] [CrossRef]
- Bull, H.J.; Hayes, S. The grpD55 locus of Escherichia coli appears to be an allele of dnaB. Mol. Gen. Genet. 1996, 252, 755–760. [Google Scholar] [PubMed]
- Hayes, S.; Erker, C.; Horbay, M.A.; Marciniuk, K.; Wang, W.; Hayes, C. Phage Lambda P Protein: Trans-Activation, Inhibition Phenotypes and their Suppression. Viruses 2013, 5, 619–653. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Duincan, D.; Hayes, C. Alcohol treatment of defective lambda lysogens is deletionogenic. Mol. Gen. Genet. 1990, 222, 17–24. [Google Scholar] [PubMed]
- Hayes, S. Mapping ethanol-induced deletions. Mol. Gen. Genet. 1991, 231, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S. Ethanol-induced genotoxicity. Mutat. Res. 1985, 143, 23–27. [Google Scholar] [CrossRef]
- Hayes, S.; Hayes, C.; Taitt, E.; Talbert, M. A simple, forward selection scheme for independently determining the toxicity and mutagenic effect of environmental chemicals: Measuring replicative killing of Escherichia coli by an integrated fragment of bacteriophage lambda DNA. In In Vitro Toxiciry Testing of Environmental Agents, Part A; Kolber, A.R., Wong, T.K., Grant, L.D., DeWoskin, R.S., Hughes, T.J., Eds.; Plenum Publishing Corp.: New York, NY, USA, 1883. [Google Scholar]
- Hayes, S.; Gordon, A.; Sadowski, I.; Hayes, C. RK bacterial test for independently measuring chemical toxicity and mutagenicity: Short-term forward selection assay. Mutat. Res. 1984, 130, 97–106. [Google Scholar] [CrossRef]
- Hayes, S.; Gordon, A. Validating RK test: Correlation with Salmonella mutatest and SOS chromotest assay results for reference compounds and influence of pH and dose response on measured toxic and mutagenic effects. Mutat. Res. 1984, 130, 107–111. [Google Scholar] [CrossRef]
- Kaiser, A.D. Lambda DNA Replication; Hershey, A.D., Ed.; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 1971. [Google Scholar]
- Rao, R.N.; Rogers, S.G. A thermoinducible lambda phage-ColE1 plasmid chimera for the overproduction of gene products from cloned DNA segments. Gene 1978, 3, 247–263. [Google Scholar] [PubMed]
- Kleckner, N. Amber mutants in the O gene of bacteriophage lambda are not efficiently complemented in the absence of phage N function. Virology 1977, 79, 174–182. [Google Scholar] [CrossRef]
- Campbell, A. Sensitive mutants of bacteriophage lambda. Virology 1961, 14, 22–32. [Google Scholar] [CrossRef]
- Hayes, S.; Wang, W.; Rajamanickam, K.; Chu, A.; Banerjee, A.; Hayes, C. Lambda gpP-DnaB Helicase Sequestration and gpP-RpoB Associated Effects: On Screens for Auxotrophs, Selection for Rif(R), Toxicity, Mutagenicity, Plasmid Curing. Viruses 2016, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Furth, M.E. Specificity Determinants for Bacteriophage Lambda DNA Replication, and Structure of the Origin of Replication; University of Wisconsin: Madison, WI, USA, 1978. [Google Scholar]
- Thomas, R.; Leurs, C.; Dambly, C.; Parmentier, D.; Lambert, L.; Brachet, P.; Lefebvre, N.; Mousset, S.; Porcheret, J.; Szpirer, J.; et al. Isolation and characterization of new sus (amber) mutants of bacteriophage lambda. Mutat. Res. 1967, 4, 735–741. [Google Scholar] [CrossRef]
- Hayes, S. Initiation of coliphage lambda replication, lit, oop RNA synthesis, and effect of gene dosage on transcription from promoters PL, PR, and PR. Virology 1979, 97, 415–438. [Google Scholar] [CrossRef]
- Furth, M.E.; Blattner, F.R.; McLeester, C.; Dove, W.F. Genetic structure of the replication origin of bacteriophage lambda. Science 1977, 198, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Asai, K.; Chu, A.M.; Hayes, C. NinR- and red-mediated phage-prophage marker rescue recombination in Escherichia coli: Recovery of a nonhomologous immlambda DNA segment by infecting lambdaimm434 phages. Genetics 2005, 170, 1485–1499. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.L.; Schroeder, J.L.; Szybalski, W.; Sanger, F.; Blattner, F.R. Appendix I. A molecular map of coliphage lambda. In Lambda II; Hendrix, R.W., Roberts, J.W., Stahl, F.W., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1983; pp. 469–517. [Google Scholar]
- Scherer, G. Nucleotide sequence of the O gene and of the origin of replication in bacteriophage lambda DNA. Nucleic Acids Res. 1978, 5, 3141–3156. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.D.; Denniston, K.; Kruger, K.E.; Furth, M.E.; Williams, B.G.; Daniels, D.L.; Blattner, F.R. Dissection and comparative anatomy of the origins of replication in lambdoid coliphages. In Proceedings of the Cold Spring Harbor Symposium Quantitative Biology; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1979; pp. 155–163. [Google Scholar]
- Zahn, K.; Blattner, F.R. Sequence-induced DNA curvature at the bacteriophage lambda origin of replication. Nature 1985, 317, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Denniston-Thompson, K.; Moore, D.D.; Kruger, K.E.; Furth, M.E.; Blattner, F.R. Physical structure of the replication origin of bacteriophage lambda. Science 1977, 198, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Zeghouf, M.; Li, J.; Butland, G.; Borkowska, A.; Canadien, V.; Richards, D.; Beattie, B.; Emili, A.; Greenblatt, J.F. Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J. Proteome Res. 2004, 3, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Hayes, C.; Bull, H.J.; Pelcher, L.A.; Slavcev, R.A. Acquired mutations in phage lambda genes O or P that enable constitutive expression of a cryptic lambdaN+cI[Ts]cro- prophage in E. coli cells shifted from 30 degreesC to 42 degreesC, accompanied by loss of immlambda and Rex+ phenotypes and emergence of a non-immune exclusion-state. Gene 1998, 223, 115–128. [Google Scholar] [PubMed]
- Tomizawa, J. Functional cooperation of genes O and P. In The Bacteriophage Lambda; Hershey, A.D., Ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1971; pp. 549–552. [Google Scholar]
- Furth, M.E.; Yates, J.L. Specificity determinants for bacteriophage lambda DNA replication. II. Structure of O proteins of lambda-phi80 and lambda-82 hybrid phages and of a lambda mutant defective in the origin of replication. J. Mol. Biol. 1978, 126, 227–240. [Google Scholar] [CrossRef]
- Wickner, S.H.; Zahn, K. Characterization of the DNA binding domain of bacteriophage lambda O protein. J. Biol. Chem. 1986, 261, 7537–7543. [Google Scholar] [PubMed]
- Gonciarz-Swiatek, M.; Wawrzynow, A.; Um, S.J.; Learn, B.A.; McMacken, R.; Kelley, W.L.; Georgopoulos, C.; Sliekers, O.; Zylicz, M. Recognition, targeting, and hydrolysis of the lambda O replication protein by the ClpP/ClpX protease. J. Biol. Chem. 1999, 274, 13999–14005. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Echols, H.; Wickner, S.; Alfano, C.; Mensa-Wilmot, K.; Gomes, B.; LeBowitz, J.; Roberts, J.D.; McMacken, R. Specialized nucleoprotein structures at the origin of replication of bacteriophage lambda: Localized unwinding of duplex DNA by a six-protein reaction. Proc. Natl. Acad. Sci. USA 1986, 83, 7638–7642. [Google Scholar] [CrossRef] [PubMed]
- Alfano, C.; McMacken, R. The role of template superhelicity in the initiation of bacteriophage lambda DNA replication. Nucleic Acids Res. 1988, 16, 9611–9630. [Google Scholar] [CrossRef] [PubMed]
- Reiser, W.; Leibrecht, I.; Klein, A. Structure and function of mutants in the P gene of bacteriophage lambda leading to the pi phenotype. Mol. Gen. Genet. 1983, 192, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Wickner, S.H. DNA replication proteins of Escherichia coli and phage lambda. Cold Spring Harb. Symp. Quant. Biol. 1979, 43, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Zylicz, M.; Gorska, I.; Taylor, K.; Georgopoulos, C. Bacteriophage lambda replication proteins: Formation of a mixed oligomer and binding to the origin of lambda DNA. Mol. Gen. Genet. 1984, 196, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Eisen, H.; Barrand, P.; Spiegelman, W.; Reichardt, L.F.; Heinemann, S.; Georgopoulos, C. Mutants in the y region of bacteriophage lambda constitutive for repressor synthesis: Their isolation and the characterization of the Hyp phenotype. Gene 1982, 20, 71–81. [Google Scholar] [CrossRef]
- Fiandt, M.; Szybalski, W.; Malamy, M.H. Polar mutations in lac, gal and phage lambda consist of a few IS-DNA sequences inserted with either orientation. Mol. Gen. Genet. 1972, 119, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Horbay, M.A.; Hayes, C. A CI-Independent Form of Replicative Inhibition: Turn Off of Early Replication of Bacteriophage Lambda. PLoS ONE 2012, 7, e36498. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Isolate | Mutated Base in λ, Mutation(s), Comment, Strain Source a |
|---|---|
| Phage lysates (#) | |
| Mutations in O | |
| λcI857 Oam905 (#1022) | 38797 G to T GAG to TAG), LT; 37 AA at N-terminal of O |
| λ [Ts] Oam29 (#51) | 38914 G to T (GAG to TAG), λ induced from AC 1966 slant R473, sc1,2; 76 AA from N-terminal of O |
| λcI857 Oam317 (#52) | 39166 G to T (GAG to TAG), LT; 160 AA at N-terminal of O |
| λ Oam8 (#50) | 39301 A to T (AAG to TAG), λ induced from AC 1966 slant R377, sc1,2; 205 AA at N-terminal of O |
| λcI857 Oam8 (#1025) | 39301 A to T (AAG to TAG), LT |
| λcI857 Oam205 (#586, 630) | 39570, C to G, (TAC to TAG), WD; 294 AA from N-terminal of O |
| Mutations in P | |
| λimm434cI Pam3 (#664) | 39786 (CAG to TAG), WD |
| λcI+prm116 Pam902 (#719,722) | 39894 (CAG to TAG), GG |
| Host b (prophage), strain # | |
| C600(λ[Ts] Oam29) #1170 | 38914 G to T (GAG to TAG) AC 1966 slant R473 (both sc’s) |
| C600(λ Oam8) #Y1169 | 39301 A to T (AAG to TAG), AC 1966 slant R377 (both sc’s) |
| C600(λ Nam7 cI857[Ind] Oam8) #Y239 | 39301 G to T (GAG to TAG), WD |
| M72 su+ (λ Nam7 cI857 r95) #Y85 c | 12 bpΔ λ bases 39123–39134, WD |
| M72 su+ (λ Nam7 cI857 r96) #Y84 c | 15 bpΔ λ bases 39139–39153, WD |
| M72 su+ (λ Nam7 cI857 r98) #Y88 | 24 bpΔ λ bases 39096–39119, WD |
| 594(λ Nam7 cI857 ti12) #188 | 39122 C to A (ACA to AAA), WD |
| Aberrant designations or with additional mutations, as received (lysate or strain #) | |
| λcI857 Oam8 (#16) | has nonsense mutation, but WT for cII-O-P-40712 in ninB; WS |
| λimm434 Oam8 (#656) | has nonsense mutation, but WT for cII-O-P-40712 in ninB; WD |
| λcI857 Oam8 (#518) | no mutation in O; 39786 in P, (CAG to TAG), same as Pam3; WS |
| λcI857 Oam29 (#582) | has nonsense mutation, WT for cII-O-P-40712 in ninB; WS |
| λcI857 Oam29 (#1023) | 38914 G to T (GAG to TAG); 38713 T to C (TTC to TCC); LT-MMS99 |
| λcI857 Oam125 (#1024) | 39511 C to T (CAG to TAG); 39182 C to T (TCC to TTC); and 39510 A to T (CAA to CAT); LT-MMS254 |
| 594(λcI857 Oam8) #Y49, Y52 | 38914 G to T (GAG to TAG), really is Oam29; WS |
| C600(λ Oam125) #1171 | 39182 C to T (TCC to TTC); and 39510 A to T (CAA to CAT); and 39511 C to T (CAG to TAG), AC 1966 slant R573 |
| Host Strains and [Plasmid] # a | EOP of λ Phage with Oam Mutants b | ||
|---|---|---|---|
| λ cI857 Oam905 c (37 AA of O) | λ cI857 Oam29 c (76 AA of O) | λ cI857 Oam205 c (294 AA of O) | |
| Pm+ SupE d | 1.0 | 1.0 | 1.0 |
| Pm− Supo | 0 | 0 | 0 |
| Complementation by O variations | |||
| O+ combinations | |||
| [O] e, p465 | 0 | 0.2 | 0.1 |
| [O-SPA] f, p472 | 0 | 1.0 | 0.6 |
| [oop-O] g, p677 | 0.05 | 0.1 | 0.1 |
| O null mutations h | |||
| [O-ilr208b], p488 | 0 | 0 | 0 |
| [O-ilr223a], p486 | 0 | 0 | 0 |
| [O-ilr541c], p485 | 0 | 0 | 0 |
| O-origin (oriλ) mutations i | |||
| [O-ori:98], p489 | 0 | 0 | 0 |
| [O-ori:95], p491 | 0.2 | 0.3 | 0.2 |
| [O-ori:96], p492 | 0.3 | 1.0 | 0.3 |
| [O-ori:ti12], p493 | 0.3 | 0.1 | 0.3 |
| O-P combinations j | |||
| [O-36P], p565 | 0 | 0.01 | 0 |
| [O-63P], p566 | 0 | 0 | 0 |
| [O-P], p569 | 0 | 0 | 0 |
| [oop-O-P] k p567, p568 | 0 | 0 | 0 |
| RK− O+ P− prophage derived from Y836 transduced into 594 | |||
| ilr 566a l | 0 | 0 | 0.002 |
| P::kan m, Bib11t | 0 | 0 | 0.001 |
| Plasmid in 594 Host Cells | Cell Viability and (Plasmid Retention per CFU Assayed; %) at CFU Growth Temperature a | |||
|---|---|---|---|---|
| 30 °C | 37 °C | 39 °C | 42 °C | |
| Only P expression | ||||
| [P] | 1.0 (35/35; 100%) | 0.01 (0/35; 0%) | 0.008 (0/35; 0%) | 0.07 (0/35; 0%) |
| dnaB-GrpD55 [P] | 1.0 (35/35; 100%) | 1.0 (35/35; 100%) | 1.0 (35/35; 100%) | 0.98 (35/35; 100%) |
| Only O expression | ||||
| [O] | 1.0 (30/30; 100%) | 1.0 (30/30; 100%) | 1.0 (30/30; 100%) | 0.61 (29/30; 97%) |
| O-P expression combinations | ||||
| [O-P] | 1.0 (62/70; 89%) | 0.12 (0/70; 0%) | 0.12 (0/70; 0%) | 0.022 (0/36; 0%) |
| [oop#1-O-P] | 1.0 (120/120; 100%) | 0.20 (98/101; 97%) | 0.005 (115/120; 96%) | 0.002 (0/36; 0%) |
| [oop#2-O-P] | 1.0 (117/120; 98%) | 0.055 (76/154; 49%) | 0.048 (62/120; 52%) | 0.008 (0/36; 0%) |
| [O-36P] | 1.0 (30/30; 100%) | 0.79 (30/30; 100%) | 0.012 (1/30; 3%) | 0.0005 (0/36; 0%) |
| [O-63P] | 1.0 (30/30; 100%) | 0.90 (30/30; 100%) | 0.055 (14/40; 35%) | 0.0023 (0/36; 0%) |
| Name | λ Map Position | Sequence (5′ to 3′) a |
|---|---|---|
| L-37904+18 | 37904–37922 | GCTGCTCTTGTGTTAATGG |
| L-MH29 | 37905–37922 | CGTCCTCAAGCTGCTCTTGTGTTAATGG |
| L20 | 39465–39484 | ACTCCGCGATAAGTGGACCC |
| L-22 | 38517–38534 | TGCTGCTTGCTGTTCTTG |
| L-PG30 | 38530–38547 | TTGGAACTGAGAAGACAG |
| L-PG1 | 38784–38801 | AAATATGCTGCTTGAGGC |
| L-38985p20 | 38985–39005 | GCAGCAAGGCGGCATGTTTGG |
| L-MH32 | 39531–39550 | CACAGATCTATAGCAAACCAAAACTCGACCTGA |
| L-18 | 39980–39996 | TTGCCGGAAGCGAGGCC |
| L-21 | 40360–40377 | CGCAACAGTAACCAGCAT |
| R-PG2 | 40747–40764 | GGTTGCGTTCCTGAATGG |
| L-Bam-O | 38686–38718 | ATATGGATCCATGACAAATACAGCAAAAATACTCAACTTCGGC |
| L-Bam-P | 39582–39606 | ATATGGATCCATGAAAAACATCGCCGCACAGATGG |
| L-Bam-OOP#1 | 38559–38580 | ATATGGATCCTGGCTCGATTGGCGCGACAAGT |
| L-Bam-OOP#2 | 38546–38577 | ATATGGATCCGTTGACGACGACATGGCTCGAT |
| L-Bam-O | 38686–38718 | ATATGGATCCATGACAAATACAGCAAAAATACTCAACTTCGGC |
| L-Bam-P | 39582–39606 | ATATGGATCCATGAAAAACATCGCCGCACAGATGG |
| R-40769m22 | 40747–40769 | GCTGCGGTTGCGTTCCTGAATGG |
| R-MH33 | 40315–40295 | GCGACGTCCCCAGGTAATGAATAATTGC |
| R-17 | 40018–40002 | TAAGACTCCGCATCCGG |
| R-MH25 | 39626–39609 | CTGCTCACGGTCAAAGTT |
| R-39280m21 | 39259–39280 | CTGCGGCGGTCAGGTCTTCTGC |
| R9+1 | 39191–39175 | TGGTCAGAGGATTCGCC |
| R-PG6 | 38569–38552 | CAATCGAGCCATGTCGTC |
| R-1536-19 | pcIpR-()-timm | GAAGACAGTCATAAGTGCGG |
| R-ClaI-P | 40280–40259 | ATATATCGATTATACACTTGCTCCTTTCAGTCCG |
| R-ClaI-O | 39582–39559 | ATATATCGATTATAGATCCACCCCGTAAATCCAGTC |
| R-ClaI-36P | 39687–39662 | ATATATCGATTACCTGCTGTACCTGCGGCTTTTCGTCG |
| R-ClaI-63P | 39768–39746 | ATATATCGATTACTTCGTTCTGGTCACGGTTAGCC |
| R-AscI-O | 39582–39559 | ATATGGCGCGCCGCTGCCGCCTAGATCCACCCCGTAAATCCAGTC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayes, S.; Rajamanickam, K.; Hayes, C. Complementation Studies of Bacteriophage ? O Amber Mutants by Allelic Forms of O Expressed from Plasmid, and O-P Interaction Phenotypes. Antibiotics 2018, 7, 31. https://doi.org/10.3390/antibiotics7020031
Hayes S, Rajamanickam K, Hayes C. Complementation Studies of Bacteriophage ? O Amber Mutants by Allelic Forms of O Expressed from Plasmid, and O-P Interaction Phenotypes. Antibiotics. 2018; 7(2):31. https://doi.org/10.3390/antibiotics7020031
Chicago/Turabian StyleHayes, Sidney, Karthic Rajamanickam, and Connie Hayes. 2018. "Complementation Studies of Bacteriophage ? O Amber Mutants by Allelic Forms of O Expressed from Plasmid, and O-P Interaction Phenotypes" Antibiotics 7, no. 2: 31. https://doi.org/10.3390/antibiotics7020031
APA StyleHayes, S., Rajamanickam, K., & Hayes, C. (2018). Complementation Studies of Bacteriophage ? O Amber Mutants by Allelic Forms of O Expressed from Plasmid, and O-P Interaction Phenotypes. Antibiotics, 7(2), 31. https://doi.org/10.3390/antibiotics7020031