Rifampicin-Loaded Mesoporous Silica Nanoparticles for the Treatment of Intracellular Infections

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
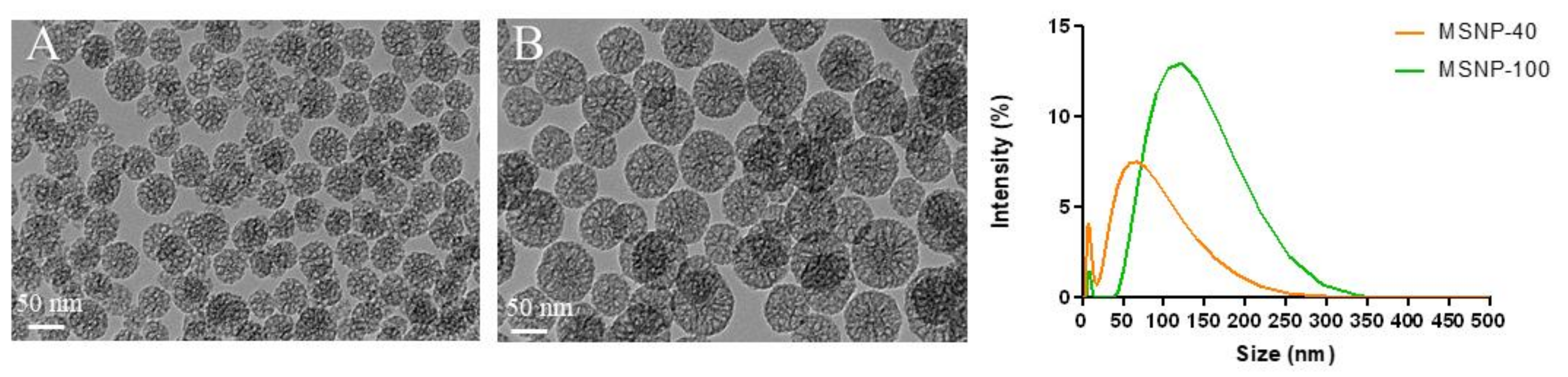
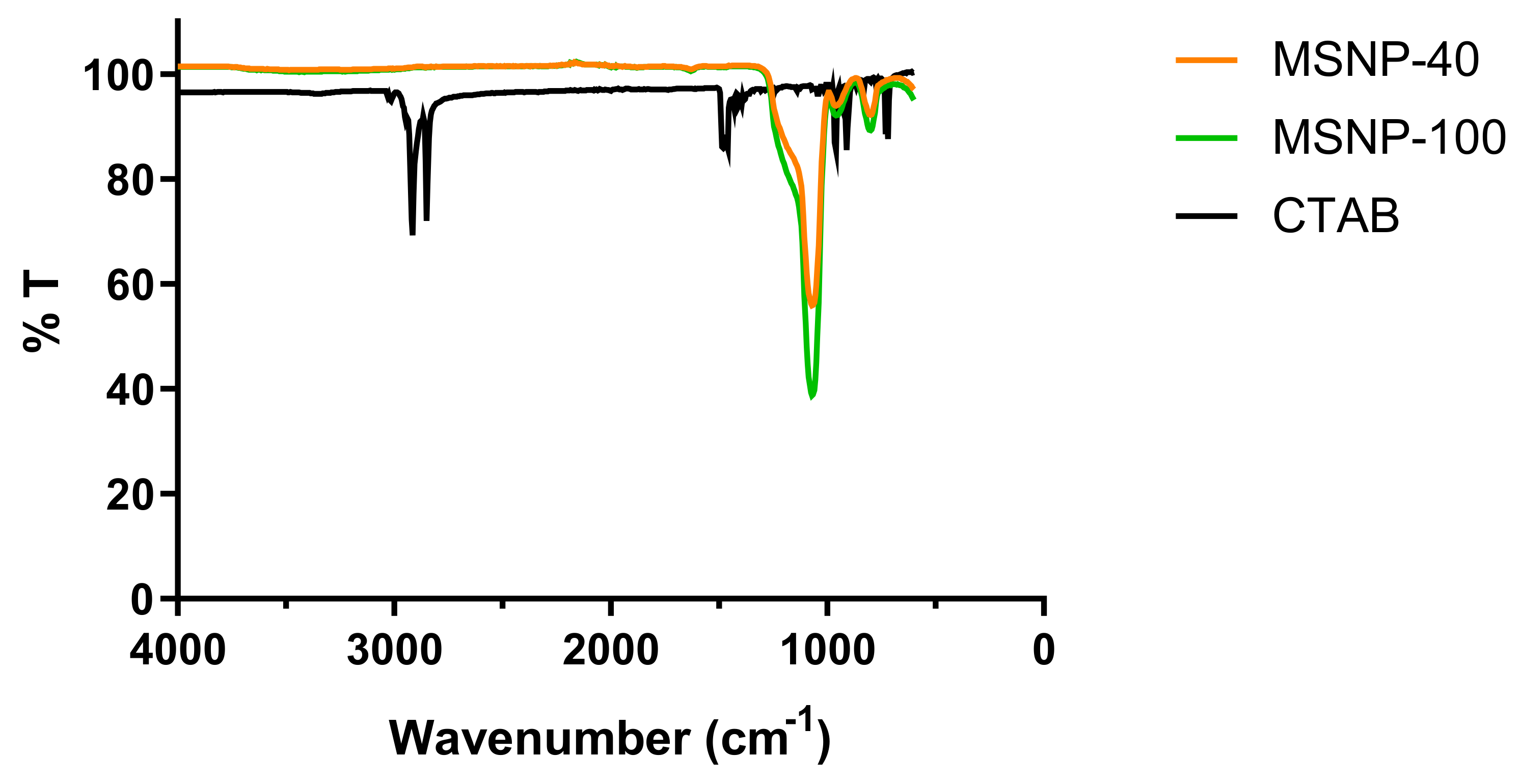
2.1. Synthesis and Characterization of MSNP
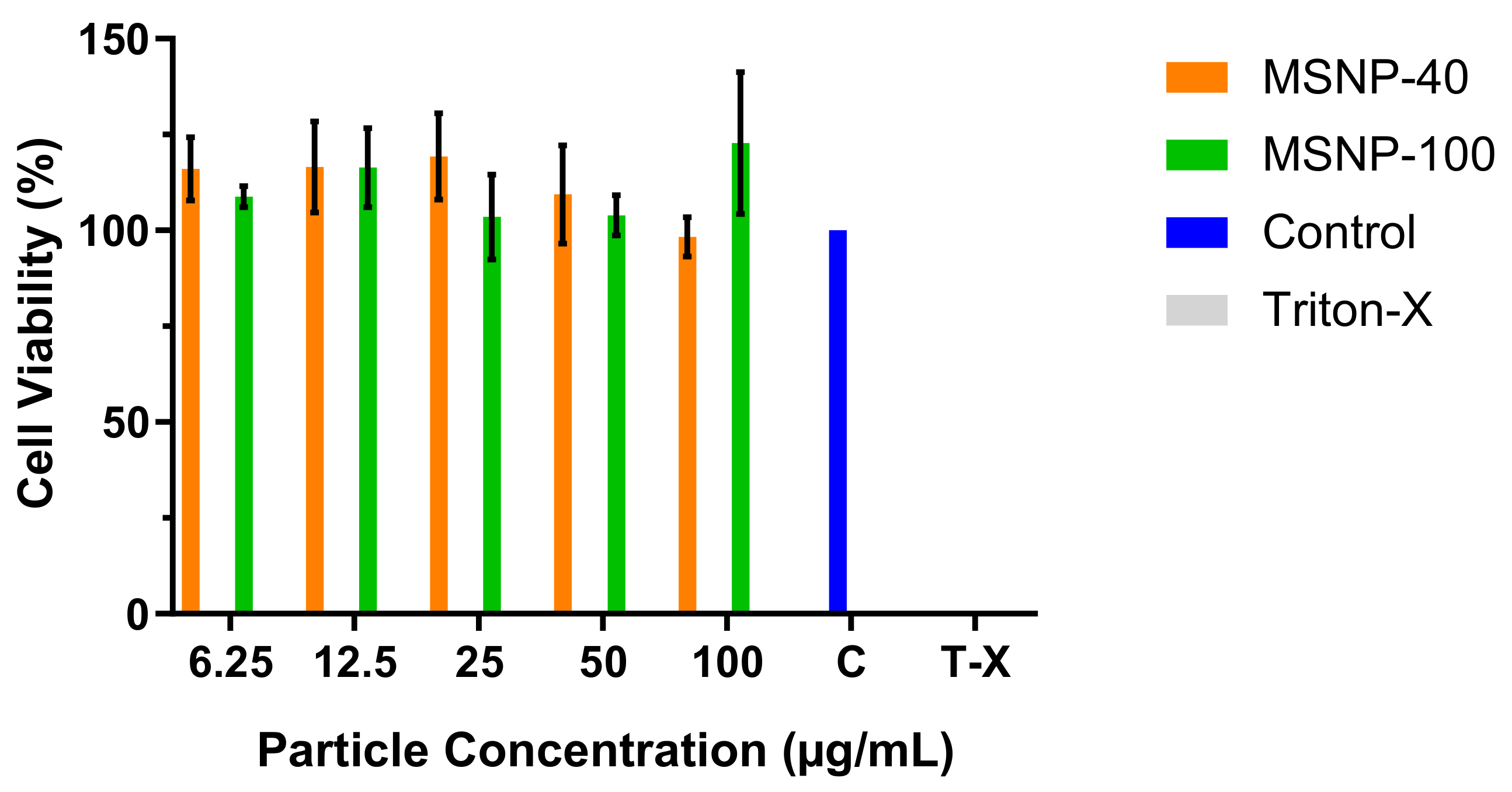
2.2. Cytotoxicity Studies
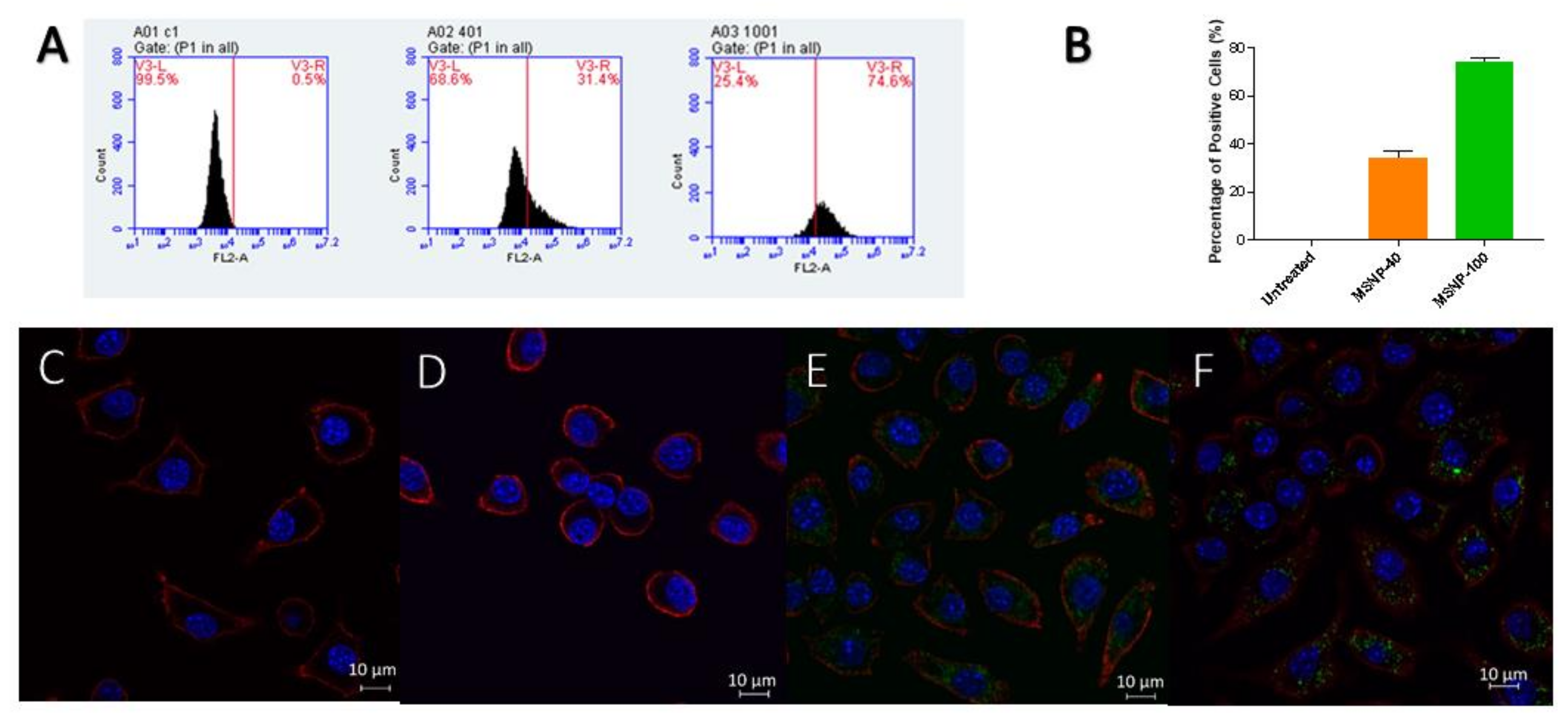
2.3. Cellular Uptake of MSNP
2.4. Loading Capacity and Encapsulation Efficiency of Rifampicin
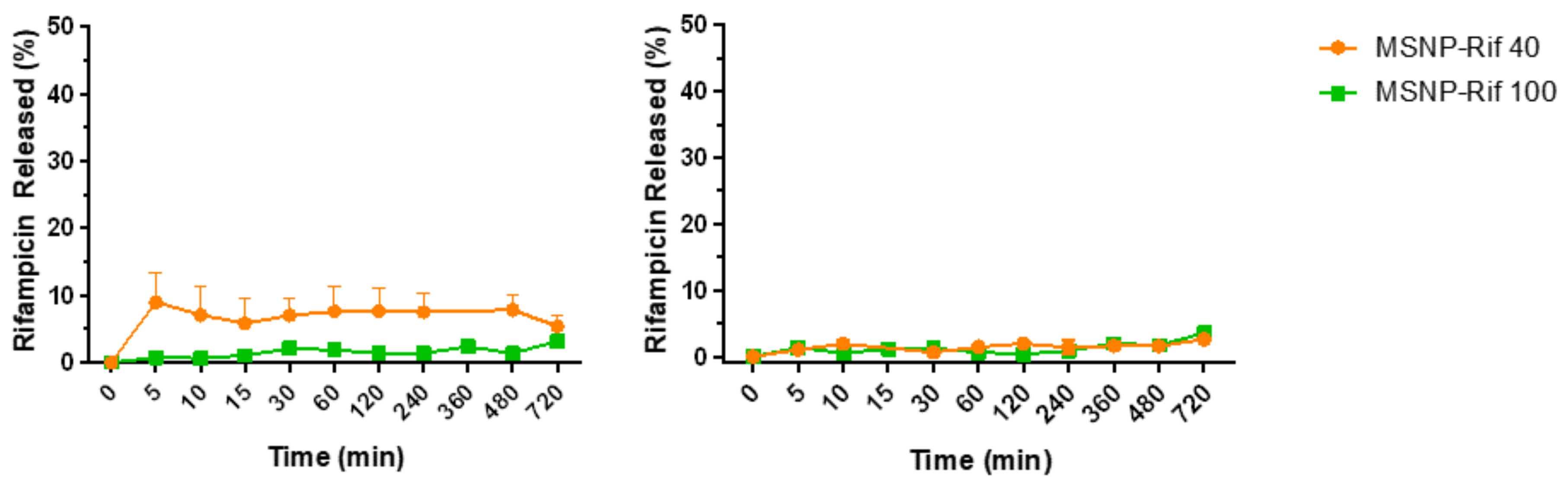
2.5. In Vitro Release Study
2.6. Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concertation (MBC) of Rifampicin in SCV S. aureus
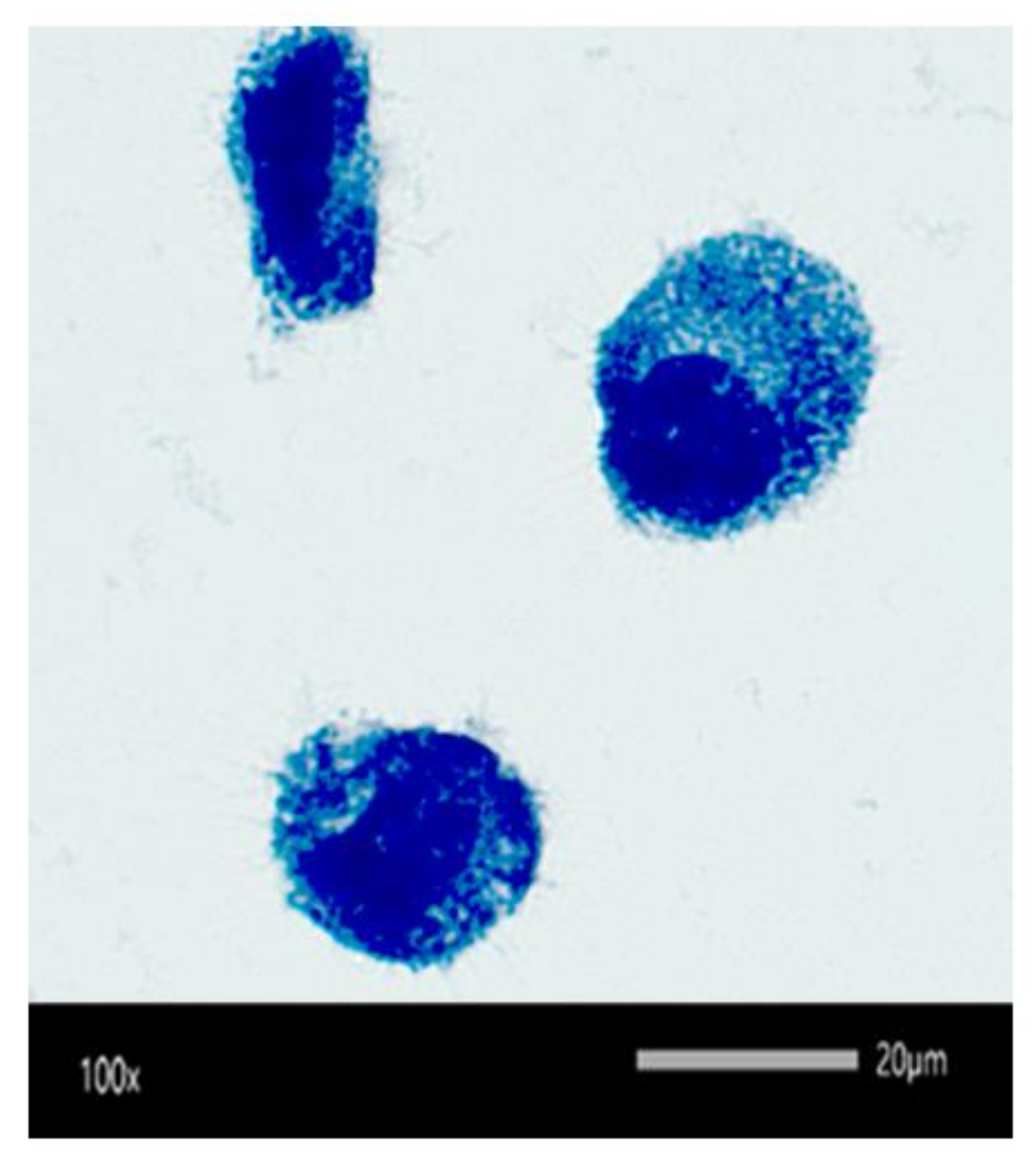
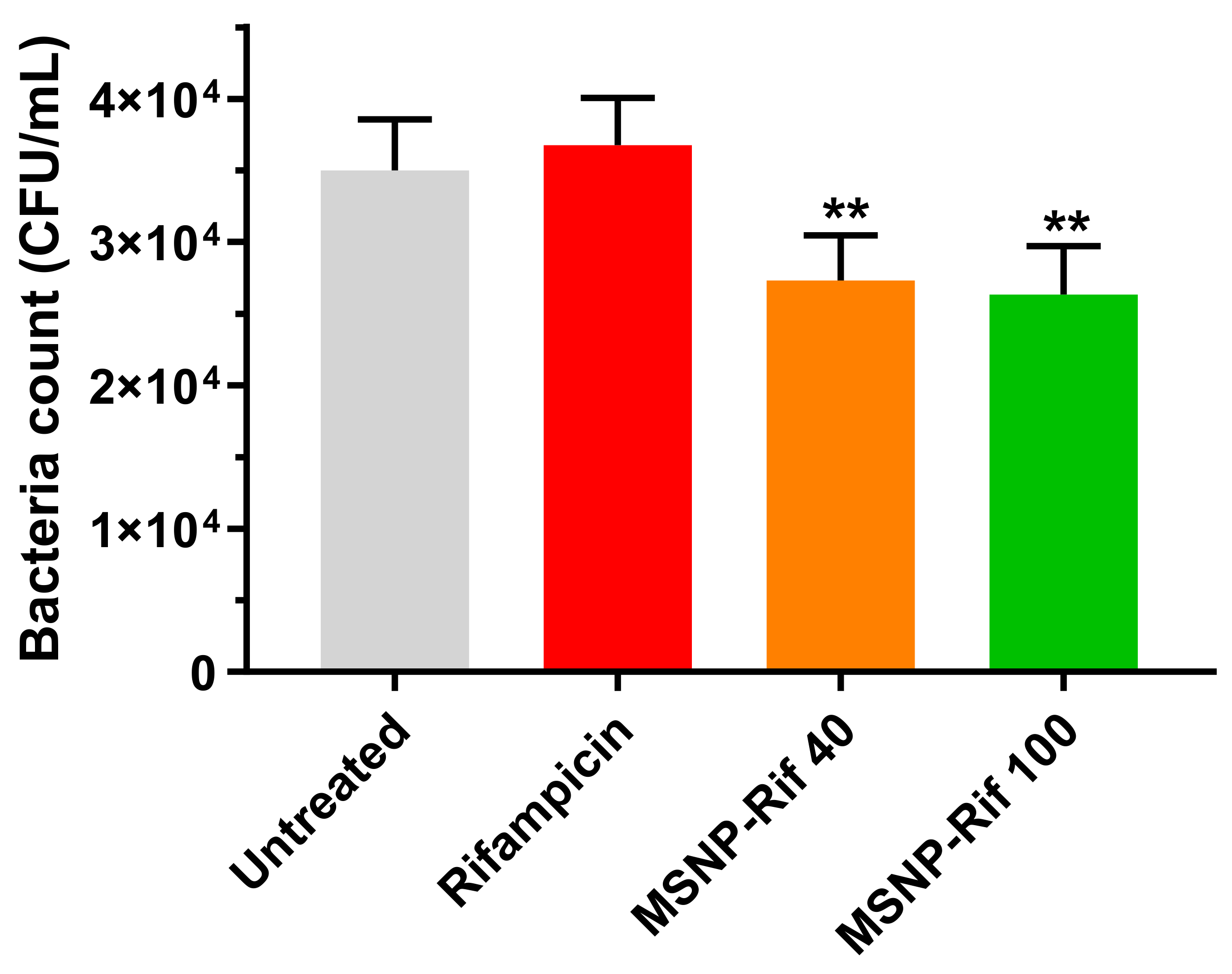
2.7. Efficacy of Rifampicin-Loaded MSNP against Intracellular SCV S. aureus
3. Materials and Methods
3.1. Materials
3.2. Fabrication of MSNP
3.3. Characterization of MSNP
3.4. Rhodamine Loading into MSNP
3.5. Cell Culture Conditions
3.6. Cytotoxicity Studies
3.7. Flow Cytometric Analysis (FACS)
3.8. Confocal Imaging
3.9. Rifampicin Loading into MSNP
3.10. In Vitro Release of Rifampicin
3.11. MIC and MBC of Rifampicin
3.12. Intracellular Infection Assay
3.13. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- America, I.D.S.O. Antimicrobial Resistance. Available online: https://www.idsociety.org/public-health/antimicrobial-resistance/antimicrobial-resistance/ (accessed on 1 March 2018).
- Kamaruzzaman, N.F.; Kendall, S.; Good, L. Targeting the hard to reach: Challenges and novel strategies in the treatment of intracellular bacterial infections. Br. J. Pharmacol. 2017, 174, 2225–2236. [Google Scholar] [CrossRef]
- Orfila, J. Definition of intracellular pathogens. Clin. Microbiol. Infect. 1996, 1, S1–S2. [Google Scholar] [CrossRef]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007. [Google Scholar] [PubMed]
- Muro, S. Drug Delivery across Physiological Barriers; Pan Stanford Publishing Pte. Ltd.: Singapore, 2016. [Google Scholar]
- Maurin, M.; Raoult, D. Optimum treatment of intracellular infection. Drugs 1996, 52, 45–59. [Google Scholar] [CrossRef]
- Tulkens, P.M. Intracellular distribution and activity of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 100–106. [Google Scholar]
- Abed, N.; Couvreur, P. Nanocarriers for antibiotics: A promising solution to treat intracellular bacterial infections. Int. J. Antimicrob. Agents 2014, 43, 485–496. [Google Scholar] [CrossRef]
- Sendi, P.; Proctor, R.A. Staphylococcus aureus as an intracellular pathogen: The role of small colony variants. Trends Microbiol. 2009, 17, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.A.; von Eiff, C.; Kahl, B.C.; Becker, K.; McNamara, P.; Herrmann, M.; Peters, G. Small colony variants: A pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 2006, 4, 295. [Google Scholar] [CrossRef]
- Proctor, R.A.; Peters, G. Small colony variants in staphylococcal infections: Diagnostic and therapeutic implications. Clin. Infect. Dis. 1998, 27, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef] [PubMed]
- Good, L.; Nielsen, P.E. Antisense inhibition of gene expression in bacteria by pna targeted to mrna. Nat. Biotechnol. 1998, 16, 355–358. [Google Scholar] [CrossRef]
- Prat, C.; Lacoma, A. Bacteria in the respiratory tract—How to treat? Or do not treat? Int. J. Infect. Dis. 2016, 51, 113–122. [Google Scholar] [CrossRef]
- Wright, L.; Rao, S.; Thomas, N.; Boulos, R.A.; Prestidge, C.A. Ramizol® encapsulation into extended release plga micro- and nanoparticle systems for subcutaneous and intramuscular administration: In vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2018, 44, 1451–1457. [Google Scholar] [CrossRef]
- Thomas, N.; Thorn, C.; Richter, K.; Thierry, B.; Prestidge, C. Efficacy of poly-lactic-co-glycolic acid micro- and nanoparticles of ciprofloxacin against bacterial biofilms. J. Pharm. Sci. 2016, 105, 3115–3122. [Google Scholar] [CrossRef]
- Sayeed, H.; Nicky, T.; Benjamin, T.; Sarah, V.; Peter-John, W.; Clive, A.P. Recent trends on the use of nanoparticles for nitric oxide delivery in antimicrobial applications. Drug Deliv. Lett. 2016, 6, 3–10. [Google Scholar]
- Imbuluzqueta, E.; Gamazo, C.; Ariza, J.; Blanco-Prieto, M.J. Drug delivery systems for potential treatment of intracellular bacterial infections. Front. Biosci. 2010, 15, 397–417. [Google Scholar] [CrossRef]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.-W.; Lin, V.S.Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef]
- Watermann, A.; Brieger, J. Mesoporous silica nanoparticles as drug delivery vehicles in cancer. Nanomaterials 2017, 7, 189. [Google Scholar] [CrossRef]
- Armstead, A.L.; Li, B. Nanomedicine as an emerging approach against intracellular pathogens. Int. J. Nanomed. 2011, 6, 3281–3293. [Google Scholar]
- Clemens, D.L.; Lee, B.-Y.; Xue, M.; Thomas, C.R.; Meng, H.; Ferris, D.; Nel, A.E.; Zink, J.I.; Horwitz, M.A. Targeted intracellular delivery of antituberculosis drugs to mycobacterium tuberculosis-infected macrophages via functionalized mesoporous silica nanoparticles. Antimicrob. Agents Chemother. 2012, 56, 2535. [Google Scholar] [CrossRef]
- Mudakavi, R.J.; Raichur, A.M.; Chakravortty, D. Lipid coated mesoporous silica nanoparticles as an oral delivery system for targeting and treatment of intravacuolar salmonella infections. Rsc Adv. 2014, 4, 61160–61166. [Google Scholar] [CrossRef]
- Mudakavi, R.J.; Vanamali, S.; Chakravortty, D.; Raichur, A.M. Development of arginine based nanocarriers for targeting and treatment of intracellular salmonella. Rsc Adv. 2017, 7, 7022–7032. [Google Scholar] [CrossRef]
- Nguyen, H.A.; Denis, O.; Vergison, A.; Theunis, A.; Tulkens, P.M.; Struelens, M.J.; Van Bambeke, F. Intracellular activity of antibiotics in a model of human thp-1 macrophages infected by a staphylococcus aureus small-colony variant strain isolated from a cystic fibrosis patient: Pharmacodynamic evaluation and comparison with isogenic normal-phenotype and revertant strains. Antimicrob. Agents Chemother. 2009, 53, 1434. [Google Scholar]
- Mohamed, W.; Sommer, U.; Sethi, S.; Domann, E.; Thormann, U.; Schutz, I.; Lips, K.; Chakraborty, T.; Schnettler, R.; Alt, V. Intracellular proliferation of s. Aureus in osteoblasts and effects of rifampicin and gentamicin on s. Aureus intracellular proliferation and survival. Eur. Cells Mater. 2014, 28, 258–268. [Google Scholar] [CrossRef]
- Perlroth, J.; Kuo, M.; Tan, J.; Bayer, A.S.; Miller, L.G. Adjunctive use of rifampin for the treatment of staphylococcus aureus infections: A systematic review of the literature. Arch. Intern. Med. 2008, 168, 805–819. [Google Scholar] [CrossRef]
- Mariappan, T.T.; Singh, S. Positioning of rifampicin in the biopharmaceutics classification system (bcs). Clin. Res. Regul. Aff. 2006, 23, 1–10. [Google Scholar] [CrossRef]
- Agrawal, S.; Panchagnula, R. Implication of biopharmaceutics and pharmacokinetics of rifampicin in variable bioavailability from solid oral dosage forms. Biopharm. Drug Dispos. 2005, 26, 321–334. [Google Scholar] [CrossRef]
- Information, N.C.f.B. Pubchem Compund Databse: Cid = 135398735. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/135398735 (accessed on 16 October 2018).
- Nandiyanto, A.B.D.; Kim, S.-G.; Iskandar, F.; Okuyama, K. Synthesis of spherical mesoporous silica nanoparticles with nanometer-size controllable pores and outer diameters. Microporous Mesoporous Mater. 2009, 120, 447–453. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhi, Z.; Jiang, T.; Zhang, J.; Wang, Z.; Wang, S. Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J. Control. Release 2010, 145, 257–263. [Google Scholar] [CrossRef]
- Lorber, B.; Fischer, F.; Bailly, M.; Roy, H.; Kern, D. Protein analysis by dynamic light scattering: Methods and techniques for students. Biochem. Mol. Biol. Educ. 2012, 40, 372–382. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676. [Google Scholar] [CrossRef]
- Quan, G.; Pan, X.; Wang, Z.; Wu, Q.; Li, G.; Dian, L.; Chen, B.; Wu, C.J.J.o.N. Lactosaminated mesoporous silica nanoparticles for asialoglycoprotein receptor targeted anticancer drug delivery. J. Nanobiotechnol. 2015, 13, 7. [Google Scholar] [CrossRef]
- Mihai, R.; Florescu, I.P.; Coroiu, V.; Oancea, A.; Lungu, M. In vitro biocompatibility testing of some synthetic polymers used for the achievement of nervous conduits. J. Med. Life 2011, 4, 250–255. [Google Scholar]
- Kuhn, D.A.; Vanhecke, D.; Michen, B.; Blank, F.; Gehr, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J. Nanotechnol. 2014, 5, 1625–1636. [Google Scholar] [CrossRef]
- Hsiao, I.L.; Mareike Gramatke, A.; Joksimovic, R.; Sokolowski, M.; Gradzielski, M.; Haase, A. Size and cell type dependent uptake of silica nanoparticles. J. Nanomed. Nanotechnol. 2014, 5, 248. [Google Scholar]
- Nabi, I.R.; Le, P.U. Caveolae/raft-dependent endocytosis. J. Cell Biol. 2003, 161, 673. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Chan, W.C.W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef]
- Almers, W. Exocytosis. Annu. Rev. Physiol. 1990, 52, 607–624. [Google Scholar] [CrossRef]
- Mohseni, M.; Gilani, K.; Mortazavi, S.A. Preparation and characterization of rifampin loaded mesoporous silica nanoparticles as a potential system for pulmonary drug delivery. Iran. J. Pharm. Res. IJPR 2015, 14, 27–34. [Google Scholar]
- Sigma-Aldrich. Rifampicin Product Information; Merck KGaA: Darmstadt, Germany, 2018. [Google Scholar]
- Sanofi-Aventis. Rifadin; Sanofi-Aventis: Bridgewater, NJ, USA, 2019. [Google Scholar]
- Bouchoucha, M.; Côté, M.-F.; C.-Gaudreault, R.; Fortin, M.-A.; Kleitz, F. Size-controlled functionalized mesoporous silica nanoparticles for tunable drug release and enhanced anti-tumoral activity. Chem. Mater. 2016, 28, 4243–4258. [Google Scholar] [CrossRef]
- Gounani, Z.; Asadollahi, M.A.; Pedersen, J.N.; Lyngsø, J.; Skov Pedersen, J.; Arpanaei, A.; Meyer, R.L. Mesoporous silica nanoparticles carrying multiple antibiotics provide enhanced synergistic effect and improved biocompatibility. Colloids Surf. B Biointerfaces 2019, 175, 498–508. [Google Scholar] [CrossRef]
- Toti, U.S.; Guru, B.R.; Hali, M.; McPharlin, C.M.; Wykes, S.M.; Panyam, J.; Whittum-Hudson, J.A. Targeted delivery of antibiotics to intracellular chlamydial infections using plga nanoparticles. Biomaterials 2011, 32, 6606–6613. [Google Scholar] [CrossRef]
- Hao, N.; Jayawardana, K.W.; Chen, X.; Yan, M. One-step synthesis of amine-functionalized hollow mesoporous silica nanoparticles as efficient antibacterial and anticancer materials. Acs Appl. Mater. Interfaces 2015, 7, 1040–1045. [Google Scholar] [CrossRef]
- Aguilar-Colomer, A.; Doadrio, J.C.; Pérez-Jorge, C.; Manzano, M.; Vallet-Regí, M.; Esteban, J. Antibacterial effect of antibiotic-loaded sba-15 on biofilm formation by staphylococcus aureus and staphylococcus epidermidis. J. Antibiot. 2016, 70, 259. [Google Scholar] [CrossRef]
- Gustafsson, H.; Isaksson, S.; Altskär, A.; Holmberg, K. Mesoporous silica nanoparticles with controllable morphology prepared from oil-in-water emulsions. J. Colloid Interface Sci. 2016, 467, 253–260. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (mic) of antimicrobial substances. Nat. Protoc. 2008, 3, 163. [Google Scholar] [CrossRef]
- Albayaty, Y.N.; Thomas, N.; Hasan, S.; Prestidge, C.A. Penetration of topically used antimicrobials through staphylococcus aureus biofilms: A comparative study using different models. J. Drug Deliv. Sci. Technol. 2018, 48, 429–436. [Google Scholar] [CrossRef]
- Bui, L.M.G.; Hoffmann, P.; Turnidge, J.D.; Zilm, P.S.; Kidd, S.P. Prolonged growth of a clinical Staphylococcus aureus strain selects for a stable small-colony-variant cell type. Infect. Immun. 2015, 83, 470. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particles | TEM (nm) | DLS (nm) (Peak Intensity) | PDI | Zeta Potential (mV) |
|---|---|---|---|---|
| MSNP-40 | 47.5 ± 4.8 | 72.9 ± 37.5 | 0.306 ± 0.01 | −20.0 ± 7.36 |
| MSNP-100 | 78.4 ± 5.7 | 130.8 ± 53.5 | 0.587 ± 0.02 | −16.9 ± 3.45 |
| Particles | Loading Capacity (% w/w) | Encapsulation Efficiency (%) |
|---|---|---|
| MSNP-Rif 40 | 38.3 | 26.8 |
| MSNP-Rif 100 | 41.1 | 22.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniam, S.; Thomas, N.; Gustafsson, H.; Jambhrunkar, M.; Kidd, S.P.; Prestidge, C.A. Rifampicin-Loaded Mesoporous Silica Nanoparticles for the Treatment of Intracellular Infections. Antibiotics 2019, 8, 39. https://doi.org/10.3390/antibiotics8020039
Subramaniam S, Thomas N, Gustafsson H, Jambhrunkar M, Kidd SP, Prestidge CA. Rifampicin-Loaded Mesoporous Silica Nanoparticles for the Treatment of Intracellular Infections. Antibiotics. 2019; 8(2):39. https://doi.org/10.3390/antibiotics8020039
Chicago/Turabian StyleSubramaniam, Santhni, Nicky Thomas, Hanna Gustafsson, Manasi Jambhrunkar, Stephen P. Kidd, and Clive A. Prestidge. 2019. "Rifampicin-Loaded Mesoporous Silica Nanoparticles for the Treatment of Intracellular Infections" Antibiotics 8, no. 2: 39. https://doi.org/10.3390/antibiotics8020039
APA StyleSubramaniam, S., Thomas, N., Gustafsson, H., Jambhrunkar, M., Kidd, S. P., & Prestidge, C. A. (2019). Rifampicin-Loaded Mesoporous Silica Nanoparticles for the Treatment of Intracellular Infections. Antibiotics, 8(2), 39. https://doi.org/10.3390/antibiotics8020039