The Continuing Threat of Methicillin-Resistant Staphylococcus aureus


Abstract
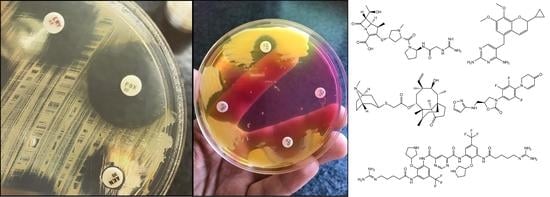
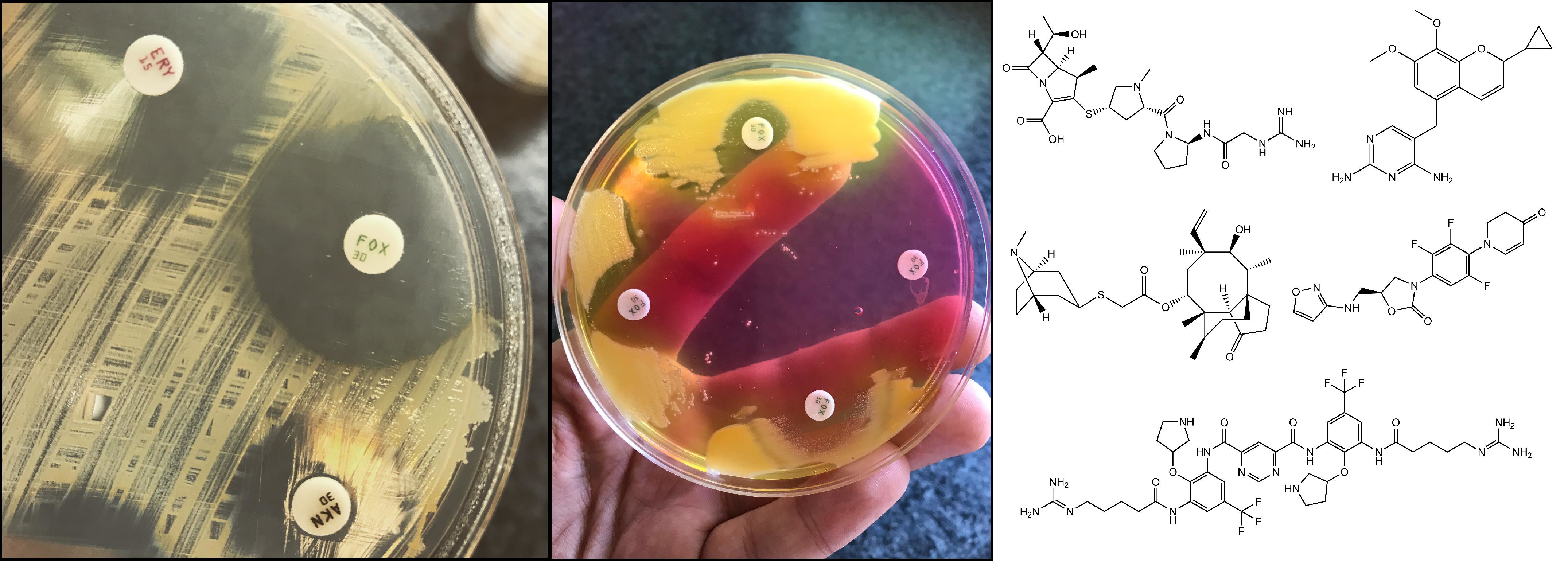
1. Introduction
2. MRSA Colonization and Screening
3. Genetics of MRSA, Typing Methods
4. Treatment Considerations, Emerging Concepts
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviations | Full |
| AST | antimicrobial susceptibility testing |
| CA | community-acquired; CAMERA |
| CAMERA | combination antibiotic therapy for methicillin-resistant Staphylococcus aureus infection (clinical trial) |
| CAP | community-acquired pneumonia |
| CC | clonal complex |
| CDC | US Centers for Disease Control and Prevention |
| cgMLST | core genome multi-locus sequence typing |
| CLSI | Clinical and Laboratory Standards Institute |
| CoNS | coagulase-negative Staphylococcus |
| CO-MRSA | community-onset MRSA |
| CRISPR/Cas9 | lustered regularly interspaced short palindromic repeats/CRISPR associated protein 9 |
| CYP | cytochrome P450 |
| DHFR | dihydrofolate-reductase |
| ESBL | extended-spectrum β-lactamase |
| ESCMID | European Society of Clinical Microbiology and Infectious Diseases |
| EU | European Union |
| EUCAST | European Committee on Antimicrobial Susceptibility Testing |
| FDA | US Food and Drug Administration |
| HA | hospital-associated |
| HAP | hospital-acquired pneumonia |
| hVISA | heterogeneous vancomycin-intermediate S. aureus |
| HLA | human leukocyte antigen |
| IAI | intra-abdominal infection |
| LA | livestock-associated |
| MALDI-TOF MS | matrix-assisted laser desorption/ionization time-of-flight mass spectrometry |
| MAO-A | monoamine-oxidase-A |
| MFS | major facilitator superfamily |
| MDR | multidrug-resistant |
| MIC | minimal inhibitory concentration |
| MLST | multi-locus sequence typing |
| MRSA | methicillin/oxacillin-resistant S. aureus |
| NGS | next-generation sequencing |
| NP | nanoparticle |
| OPAT | outpatient parenteral antibiotic therapy |
| PFGE | pulse-field gel electrophoresis |
| PBP | penicillin-binding protein |
| QS | quorum sensing |
| UTI | urinary tract infection |
| PDI | prosthetic device infection |
| PCR | polymerase chain reaction |
| PVL | Panton–Valentine leucocidin |
| QRDR | quinolone resistance-determining region |
| QTc | corrected QT-interval |
| RNA | ribonucleic acid |
| RNAi | RNA-interference |
| SCV | small-colony variant |
| SMX/TMP | co-trimoxazole |
| SSTI | skin and soft tissue infection |
| ST | sequence type |
| ssDNA | single-strand DNA |
| ssRNA | single-strand RNA |
| TDM | therapeutic drug monitoring |
| TAT | turnaround time |
| TLR | toll-like receptor |
| TSS | toxic shock syndrome |
| TSST | toxic shock syndrome toxin |
| UTI | urinary tract infection |
| VAP | ventilator-associated pneumonia |
| VISA | vancomycin-intermediate S. aureus |
| VNTR | variable number tandem repeat |
| VRE | vancomycin-resistant enterococci |
| VRSA | vancomycin-resistant S. aureus |
| WGS | whole-genome sequencing |
References
- Murray, P.R.; Baron, E.J.; Jorgensen, J.H.; Landry, M.L.; Pfaller, M.A. Manual of Clinical Microbiology, 9th ed.; American Society for Microbiology: Washington, DC, USA, 2007; ISBN 978-1-55581-371-0. [Google Scholar]
- Pulverer, G. Taxonomy of Staphylococcus aureus. Zentralbl. Bakteriol. Mikrobiol. Hyg. A. 1986, 262, 425–437. [Google Scholar] [CrossRef]
- Shaw, C.; Stitt, J.M.; Cowan, S.T. Staphylococci and their Classification. J. Gen. Microbiol. 1951, 5, 1010–1023. [Google Scholar] [CrossRef]
- Melter, O.; Radojevič, B. Small colony variants of Staphylococcus aureus--review. Folia Microbiol. (Praha) 2010, 55, 548–558. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-Negative Staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef]
- Gould, D.; Chamberlaine, A. Staphylococcus aureus: A review of the literature. J. Clin. Nurs. 1995, 4, 5–12. [Google Scholar] [CrossRef]
- Garcia, L.G.; Lemaire, S.; Kahl, B.C.; Becker, K.; Proctor, R.A.; Denis, O.; Tulkens, P.M.; Van Bambeke, F. Antibiotic activity against small-colony variants of Staphylococcus aureus: Review of in vitro, animal and clinical data. J. Antimicrob. Chemother. 2013, 68, 1455–1464. [Google Scholar] [CrossRef]
- Kahl, B.C.; Becker, K.; Löffler, B. Clinical Significance and Pathogenesis of Staphylococcal Small Colony Variants in Persistent Infections. Clin. Microbiol. Rev. 2016, 29, 401–427. [Google Scholar] [CrossRef]
- Proctor, R.A.; Kriegeskorte, A.; Kahl, B.C.; Becker, K.; Löffler, B.; Peters, G. Staphylococcus aureus Small Colony Variants (SCVs): A road map for the metabolic pathways involved in persistent infections. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef]
- Menetrey, A.; Janin, A.; Pullman, J.; Overcash, J.S.; Haouala, A.; Leylavergne, F.; Turbe, L.; Wittke, F.; Nicolas-Métral, V. Bone and Joint Tissue Penetration of the Staphylococcus-Selective Antibiotic Afabicin in Patients Undergoing Elective Hip Replacement Surgery. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Kang, C.-I.; Song, J.-H.; Ko, K.S.; Chung, D.R.; Peck, K.R. Clinical features and outcome of Staphylococcus aureus infection in elderly versus younger adult patients. Int. J. Infect. Dis. 2011, 15, e58–e62. [Google Scholar] [CrossRef]
- Kobayashi, S.D.; Malachowa, N.; DeLeo, F.R. Pathogenesis of Staphylococcus aureus Abscesses. Am. J. Pathol. 2015, 185, 1518–1527. [Google Scholar] [CrossRef]
- Gijón, M.; Bellusci, M.; Petraitiene, B.; Noguera-Julian, A.; Zilinskaite, V.; Sanchez Moreno, P.; Saavedra-Lozano, J.; Glikman, D.; Daskalaki, M.; Kaiser-Labusch, P.; et al. Factors associated with severity in invasive community-acquired Staphylococcus aureus infections in children: A prospective European multicentre study. Clin. Microbiol. Infect. 2016, 22, 643.e1–643.e6. [Google Scholar] [CrossRef]
- Ericson, J.E.; Popoola, V.O.; Smith, P.B.; Benjamin, D.K.; Fowler, V.G.; Benjamin, D.K.; Clark, R.H.; Milstone, A.M. Burden of Invasive Staphylococcus aureus Infections in Hospitalized Infants. JAMA Pediatr. 2015, 169, 1105–1111. [Google Scholar] [CrossRef]
- Wang, L.J.; Dong, F.; Qian, S.Y.; Yao, K.H.; Song, W.Q. Clinical and Molecular Epidemiology of Invasive Staphylococcus aureus Infections in Chinese Children: A Single-center Experience. Chin. Med. J. (Engl.) 2017, 130, 2889–2890. [Google Scholar] [CrossRef]
- Powers, M.E.; Wardenburg, J.B. Igniting the Fire: Staphylococcus aureus Virulence Factors in the Pathogenesis of Sepsis. PLoS Pathog. 2014, 10, e1003871. [Google Scholar] [CrossRef]
- Oogai, Y.; Matsuo, M.; Hashimoto, M.; Kato, F.; Sugai, M.; Komatsuzawa, H. Expression of Virulence Factors by Staphylococcus aureus Grown in Serum. Appl. Environ. Microbiol. 2011, 77, 8097–8105. [Google Scholar] [CrossRef]
- Silversides, J.A.; Lappin, E.; Ferguson, A.J. Staphylococcal toxic shock syndrome: Mechanisms and management. Curr. Infect. Dis. Rep. 2010, 12, 392–400. [Google Scholar] [CrossRef]
- Kumar, A.; Tassopoulos, A.M.; Li, Q.; Yu, F.-S.X. Staphylococcus aureus protein A induced inflammatory response in human corneal epithelial cells. Biochem. Biophys. Res. Commun. 2007, 354, 955–961. [Google Scholar] [CrossRef]
- Lacey, K.A.; Geoghegan, J.A.; McLoughlin, R.M. The Role of Staphylococcus aureus Virulence Factors in Skin Infection and Their Potential as Vaccine Antigens. Pathogens 2016, 5. [Google Scholar] [CrossRef]
- Gomes-Fernandes, M.; Laabei, M.; Pagan, N.; Hidalgo, J.; Molinos, S.; Villar Hernandez, R.; Domínguez-Villanueva, D.; Jenkins, A.T.A.; Lacoma, A.; Prat, C. Accessory gene regulator (Agr) functionality in Staphylococcus aureus derived from lower respiratory tract infections. PLoS ONE 2017, 12, e0175552. [Google Scholar] [CrossRef]
- Haque, S.; Ahmad, F.; Dar, S.A.; Jawed, A.; Mandal, R.K.; Wahid, M.; Lohani, M.; Khan, S.; Singh, V.; Akhter, N. Developments in strategies for Quorum Sensing virulence factor inhibition to combat bacterial drug resistance. Microb. Pathog. 2018, 121, 293–302. [Google Scholar] [CrossRef]
- Seidl, K.; Stucki, M.; Ruegg, M.; Goerke, C.; Wolz, C.; Harris, L.; Berger-Bächi, B.; Bischoff, M. Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob. Agents Chemother. 2006, 50, 1183–1194. [Google Scholar] [CrossRef]
- Erdem, H.; Tetik, A.; Arun, O.; Besirbellioglu, B.A.; Coskun, O.; Eyigun, C.P. War and infection in the pre-antibiotic era: The Third Ottoman Army in 1915. Scand. J. Infect. Dis. 2011, 43, 690–695. [Google Scholar] [CrossRef]
- Gaynes, R. The Discovery of Penicillin—New Insights After More Than 75 Years of Clinical Use. Emerg. Infect. Dis. 2017, 23, 849–853. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Papanicolas, L.E.; Bell, J.M.; Bastian, I. Performance of phenotypic tests for detection of penicillinase in Staphylococcus aureus isolates from Australia. J. Clin. Microbiol. 2014, 52, 1136–1138. [Google Scholar] [CrossRef]
- Holten, K.B.; Onusko, E.M. Appropriate Prescribing of Oral Beta-Lactam Antibiotics. AFP 2000, 62, 611–620. [Google Scholar]
- Lobanovska, M.; Pilla, G. Penicillin’s Discovery and Antibiotic Resistance: Lessons for the Future? Yale J. Biol. Med. 2017, 90, 135–145. [Google Scholar]
- Eady, E.A.; Cove, J.H. Staphylococcal resistance revisited: Community-acquired methicillin resistant Staphylococcus aureus—An emerging problem for the management of skin and soft tissue infections. Curr. Opin. Infect. Dis. 2003, 16, 103–124. [Google Scholar] [CrossRef]
- Sabath, L.D.; Finland, M. Inactivation of methicillin, oxacillin and ancillin by Staphylococcus aureus. Proc. Soc. Exp. Biol. Med. 1962, 111, 547–550. [Google Scholar] [CrossRef]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203. [Google Scholar] [CrossRef]
- Mylonas, I. Antibiotic chemotherapy during pregnancy and lactation period: Aspects for consideration. Arch. Gynecol. Obstet. 2011, 283, 7–18. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- van Duin, D.; Paterson, D. Multidrug Resistant Bacteria in the Community: Trends and Lessons Learned. Infect. Dis. Clin. N. Am. 2016, 30, 377–390. [Google Scholar] [CrossRef]
- Kaur, D.C.; Chate, S.S. Study of Antibiotic Resistance Pattern in Methicillin Resistant Staphylococcus Aureus with Special Reference to Newer Antibiotic. J. Glob. Infect. Dis. 2015, 7, 78–84. [Google Scholar] [CrossRef]
- Gajdács, M.; Paulik, E.; Szabó, A. [The opinions of community pharmacists related to antibiotic use and resistance] (article in Hungarian). Acta Pharm. Hung. 2018, 88, 249–252. [Google Scholar]
- Gajdács, M.; Paulik, E.; Szabó, A. [The attitude of community pharmacists towards their widening roles in the prevention and treatment of infectious diseases in the southeast region of Hungary] (article in Hungarian). Gyógyszerészet 2019, 63, 26–30. [Google Scholar]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed. Res. Int. 2016, 2016. [Google Scholar] [CrossRef]
- Dyar, O.J.; Huttner, B.; Schouten, J.; Pulcini, C. What is antimicrobial stewardship? Clin. Microbiol. Infect. 2017, 23, 793–798. [Google Scholar] [CrossRef]
- Ha, D.R.; Haste, N.M.; Gluckstein, D.P. The Role of Antibiotic Stewardship in Promoting Appropriate Antibiotic Use. Am. J. Lifestyle Med. 2017. [Google Scholar] [CrossRef]
- Bergeron, J. Prudent use of antibiotics. Can. Vet. J. 2014, 55, 714. [Google Scholar]
- Phillips, I. Prudent Use of Antibiotics: Are Our Expectations Justified? Clin. Infect. Dis. 2001, 33, S130–S132. [Google Scholar] [CrossRef]
- Lee, B.Y.; Singh, A.; David, M.Z.; Bartsch, S.M.; Slayton, R.B.; Huang, S.S.; Zimmer, S.M.; Potter, M.A.; Macal, C.M.; Lauderdale, D.S.; et al. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin. Microbiol. Infect. 2013, 19, 528–536. [Google Scholar] [CrossRef]
- Gajdács, M. Extra deaths due to pandrug resistant bacteria: A survey of the literature. Egészségfejlesztés 2019, 60, 31–36. [Google Scholar]
- Cosgrove, S.E.; Sakoulas, G.; Perencevich, E.N.; Schwaber, M.J.; Karchmer, A.W.; Carmeli, Y. Comparison of Mortality Associated with Methicillin-Resistant and Methicillin-Susceptible Staphylococcus aureus Bacteremia: A Meta-analysis. Clin. Infect. Dis. 2003, 36, 53–59. [Google Scholar] [CrossRef] [PubMed]
- van Hal, S.J.; Jensen, S.O.; Vaska, V.L.; Espedido, B.A.; Paterson, D.L.; Gosbell, I.B. Predictors of Mortality in Staphylococcus aureus Bacteremia. Clin. Microbiol. Rev. 2012, 25, 362–386. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Salata, R.A.; Bonomo, R.A. Current and novel antibiotics against resistant Gram-positive bacteria. Infect. Drug Resist. 2008, 1, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, A.; Linden, P.K.; Friedman, B. Incidence, prevalence, and management of MRSA bacteremia across patient populations—A review of recent developments in MRSA management and treatment. Crit. Care 2017, 21. [Google Scholar] [CrossRef] [PubMed]
- Graffunder, E.M.; Venezia, R.A. Risk factors associated with nosocomial methicillin-resistant Staphylococcus aureus (MRSA) infection including previous use of antimicrobials. J. Antimicrob. Chemother. 2002, 49, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Rosendal, K.; Jessen, O. Epidemic spread of Staphylococcus aureus PHAGE-TYPE 83A. Acta Pathol. Microbiol. Scand. 1964, 60, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Enright, M.C.; Robinson, D.A.; Randle, G.; Feil, E.J.; Grundmann, H.; Spratt, B.G. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. USA 2002, 99, 7687–7692. [Google Scholar] [CrossRef] [PubMed]
- Morell, E.A.; Balkin, D.M. Methicillin-Resistant Staphylococcus Aureus: A Pervasive Pathogen Highlights the Need for New Antimicrobial Development. Yale J. Biol. Med. 2010, 83, 223–233. [Google Scholar]
- David, M.Z.; Daum, R.S. Community-associated methicillin-resistant Staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 2010, 23, 616–687. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B. Five decades of MRSA: Controversy and uncertainty continues. Lancet 2011, 378, 1291–1292. [Google Scholar] [CrossRef]
- King, M.D.; Humphrey, B.J.; Wang, Y.F.; Kourbatova, E.V.; Ray, S.M.; Blumberg, H.M. Emergence of community-acquired methicillin-resistant Staphylococcus aureus USA 300 clone as the predominant cause of skin and soft-tissue infections. Ann. Intern. Med. 2006, 144, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.C. MRSA: The first half century. J. Antimicrob. Chemother. 2012, 67, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J.V. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014, 22, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A. Mechanisms of Resistance to Quinolones. Clin. Infect. Dis. 2005, 41, S120–S126. [Google Scholar] [CrossRef]
- Dukic, V.M.; Lauderdale, D.S.; Wilder, J.; Daum, R.S.; David, M.Z. Epidemics of Community-Associated Methicillin-Resistant Staphylococcus aureus in the United States: A Meta-Analysis. PLoS ONE 2013, 8, e52722. [Google Scholar] [CrossRef]
- Chapman, A.L.N. Outpatient parenteral antimicrobial therapy. BMJ 2013, 346, f1585. [Google Scholar] [CrossRef] [PubMed]
- Laupland, K.B.; Valiquette, L. Outpatient parenteral antimicrobial therapy. Can. J. Infect. Dis. Med. Microbiol. 2013, 24, 9–11. [Google Scholar] [CrossRef]
- Peton, V.; Le Loir, Y. Staphylococcus aureus in veterinary medicine. Infect. Genet. Evol. 2014, 21, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Cuny, C.; Wieler, L.H.; Witte, W. Livestock-Associated MRSA: The Impact on Humans. Antibiotics (Basel) 2015, 4, 521–543. [Google Scholar] [CrossRef] [PubMed]
- Cuny, C.; Köck, R.; Witte, W. Livestock associated MRSA (LA-MRSA) and its relevance for humans in Germany. Int. J. Med. Microbiol. 2013, 303, 331–337. [Google Scholar] [CrossRef]
- Dorado-García, A.; Bos, M.E.; Graveland, H.; Cleef, B.A.V.; Verstappen, K.M.; Kluytmans, J.A.; Wagenaar, J.A.; Heederik, D.J. Risk factors for persistence of livestock-associated MRSA and environmental exposure in veal calf farmers and their family members: An observational longitudinal study. BMJ Open 2013, 3, e003272. [Google Scholar] [CrossRef]
- Sharma, M.; Nunez-Garcia, J.; Kearns, A.M.; Doumith, M.; Butaye, P.R.; Argudín, M.A.; Lahuerta-Marin, A.; Pichon, B.; AbuOun, M.; Rogers, J.; et al. Livestock-Associated Methicillin Resistant Staphylococcus aureus (LA-MRSA) Clonal Complex (CC) 398 Isolated from UK Animals belong to European Lineages. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Kevorkijan, B.K.; Petrovič, Ž.; Kocuvan, A.; Rupnik, M. MRSA diversity and the emergence of LA-MRSA in a large teaching hospital in Slovenia. Acta Microbiol. Immunol. Hung. 2019, 1–12. [Google Scholar] [CrossRef]
- Butaye, P.; Argudín, M.A.; Smith, T.C. Livestock-Associated MRSA and Its Current Evolution. Curr. Clin. Microbiol. Rep. 2016, 3, 19–31. [Google Scholar] [CrossRef]
- Gajdács, M.; Spengler, G.; Urbán, E. Identification and Antimicrobial Susceptibility Testing of Anaerobic Bacteria: Rubik’s Cube of Clinical Microbiology? Antibiotics 2017, 6, 25. [Google Scholar] [CrossRef]
- Lawson, P.A.; Citron, D.M.; Tyrrell, K.L.; Finegold, S.M. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prevot 1938. Anaerobe 2016, 40, 95–99. [Google Scholar] [CrossRef]
- Khan, F.Y.; Elzouki, A.-N. Clostridium difficile infection: A review of the literature. Asian Pac. J. Trop. Med. 2014, 7, S6–S13. [Google Scholar] [CrossRef]
- MacFadden, D.R.; Lipsitch, M.; Olesen, S.W.; Grad, Y. Multidrug-resistant Neisseria gonorrhoeae: Implications for future treatment strategies. Lancet Infect. Dis. 2018, 18, 599. [Google Scholar] [CrossRef]
- Rupp, M.E.; Fey, P.D. Extended spectrum beta-lactamase (ESBL)-producing Enterobacteriaceae: Considerations for diagnosis, prevention and drug treatment. Drugs 2003, 63, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Paterson, D.L. Carbapenemase-Producing Enterobacteriaceae. Semin. Respir. Crit. Care Med. 2015, 36, 74–84. [Google Scholar] [PubMed]
- CDC The biggest antibiotic-resistant threats in the U.S. Available online: https://www.cdc.gov/drugresistance/biggest_threats.html (accessed on 27 March 2019).
- Nasim, J.; Witek, K.; Kincses, A.; Abdin, A.Y.; Żesławska, E.; Marć, M.A.; Gajdács, M.; Spengler, G.; Nitek, W.; Latacz, G.; et al. Pronounced activity of aromatic selenocyanates against multidrug resistant ESKAPE bacteria. New J. Chem. 2019, 15. [Google Scholar] [CrossRef]
- Abbas, M.; Paul, M.; Huttner, A. New and improved? A review of novel antibiotics for Gram-positive bacteria. Clin. Microbiol. Infect. 2017, 23, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Young, K.; Silver, L.L. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef]
- Campanini-Salinas, J.; Andrades-Lagos, J.; Mella-Raipan, J.; Vasquez-Velasquez, D. Novel Classes of Antibacterial Drugs in Clinical Development, a Hope in a Post-antibiotic Era. Curr. Top. Med. Chem. 2018, 18, 1188–1202. [Google Scholar] [CrossRef]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug Resistant Bacterial Infections, Including Tuberculosis. Available online: http://www.who.int/medicines/areas/rational_use/prioritization-of-pathogens/en/ (accessed on 27 March 2019).
- Stefani, S.; Chung, D.R.; Lindsay, J.A.; Friedrich, A.W.; Kearns, A.M.; Westh, H.; Mackenzie, F.M. Meticillin-resistant Staphylococcus aureus (MRSA): Global epidemiology and harmonisation of typing methods. Int. J. Antimicrob. Agents 2012, 39, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.M.; Hennessy, T.; Baggett, H.C.; Bruden, D.; Parks, D.; Klejka, J. Methicillin-Resistant Staphylococcus aureus Carriage and Risk Factors for Skin Infections, Southwestern Alaska, USA. Emerg. Infect. Dis. 2010, 16, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Sollid, J.U.E.; Furberg, A.S.; Hanssen, A.M.; Johannessen, M. Staphylococcus aureus: Determinants of human carriage. Infect. Genet. Evol. 2014, 21, 531–541. [Google Scholar] [CrossRef]
- McConeghy, K.W.; Mikolich, D.J.; LaPlante, K.L. Agents for the decolonization of methicillin-resistant Staphylococcus aureus. Pharmacotherapy 2009, 29, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.F.; Liao, C.H.; Pai, M.F.; Chu, F.Y.; Hsu, S.P.; Chen, H.Y.; Yang, J.Y.; Chiu, Y.L.; Peng, Y.S.; Chang, S.C.; et al. Nasal Carriage of Methicillin-resistant Staphylococcus aureus Is Associated with Higher All-Cause Mortality in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 167–174. [Google Scholar] [CrossRef]
- Dulon, M.; Peters, C.; Schablon, A.; Nienhaus, A. MRSA carriage among healthcare workers in non-outbreak settings in Europe and the United States: A systematic review. BMC Infect. Dis. 2014, 14, 363. [Google Scholar] [CrossRef]
- Safdar, N.; Narans, L.; Gordon, B.; Maki, D.G. Comparison of Culture Screening Methods for Detection of Nasal Carriage of Methicillin-Resistant Staphylococcus aureus: A Prospective Study Comparing 32 Methods. J. Clin. Microbiol. 2003, 41, 3163–3166. [Google Scholar] [CrossRef]
- Ábrók, M.; Lázár, A.; Szécsényi, M.; Deák, J.; Urbán, E. Combination of MALDI-TOF MS and PBP2’ latex agglutination assay for rapid MRSA detection. J. Microbiol. Methods 2018, 144, 122–124. [Google Scholar] [CrossRef]
- Wolk, D.M.; Marx, J.L.; Dominguez, L.; Driscoll, D.; Schifman, R.B. Comparison of MRSASelect Agar, CHROMagar Methicillin-Resistant Staphylococcus aureus (MRSA) Medium, and Xpert MRSA PCR for Detection of MRSA in Nares: Diagnostic Accuracy for Surveillance Samples with Various Bacterial Densities. J. Clin. Microbiol. 2009, 47, 3933–3936. [Google Scholar] [CrossRef]
- Leclercq, R.; Cantón, R.; Brown, D.F.J.; Giske, C.G.; Heisig, P.; MacGowan, A.P.; Mouton, J.W.; Nordmann, P.; Rodloff, A.C.; Rossolini, G.M.; et al. EUCAST expert rules in antimicrobial susceptibility testing. Clin. Microbiol. Infect. 2013, 19, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute (CLSI). Available online: https://clsi.org/standards/products/microbiology/ (accessed on 10 September 2017).
- Broekema, N.M.; Van, T.T.; Monson, T.A.; Marshall, S.A.; Warshauer, D.M. Comparison of Cefoxitin and Oxacillin Disk Diffusion Methods for Detection of mecA-Mediated Resistance in Staphylococcus aureus in a Large-Scale Study. J. Clin. Microbiol. 2009, 47, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Wolk, D.M.; Struelens, M.J.; Pancholi, P.; Davis, T.; Della-Latta, P.; Fuller, D.; Picton, E.; Dickenson, R.; Denis, O.; Johnson, D.; et al. Rapid Detection of Staphylococcus aureus and Methicillin-Resistant S. aureus (MRSA) in Wound Specimens and Blood Cultures: Multicenter Preclinical Evaluation of the Cepheid Xpert MRSA/SA Skin and Soft Tissue and Blood Culture Assays. J. Clin. Microbiol. 2009, 47, 823–826. [Google Scholar] [CrossRef]
- Benagli, C.; Rossi, V.; Dolina, M.; Tonolla, M.; Petrini, O. Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry for the Identification of Clinically Relevant Bacteria. PLoS ONE 2011, 6, e16424. [Google Scholar] [CrossRef] [PubMed]
- Shore, A.C.; Deasy, E.C.; Slickers, P.; Brennan, G.; O’Connell, B.; Monecke, S.; Ehricht, R.; Coleman, D.C. Detection of staphylococcal cassette chromosome mec type XI carrying highly divergent mecA, mecI, mecR1, blaZ, and ccr genes in human clinical isolates of clonal complex 130 methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 3765–3773. [Google Scholar] [CrossRef]
- Murray, B.E. The life and times of the Enterococcus. Clin. Microbiol. Rev. 1990, 3, 46–65. [Google Scholar] [CrossRef]
- Hakenbeck, R. Transformation in Streptococcus pneumoniae: Mosaic genes and the regulation of competence. Res. Microbiol. 2000, 151, 453–456. [Google Scholar] [CrossRef]
- Deurenberg, R.H.; Stobberingh, E.E. The evolution of Staphylococcus aureus. Infect. Genet. Evol. 2008, 8, 747–763. [Google Scholar] [CrossRef]
- Ito, T.; Kuwahara-Arai, K.; Katayama, Y.; Uehara, Y.; Han, X.; Kondo, Y.; Hiramatsu, K. Staphylococcal Cassette Chromosome mec (SCCmec) analysis of MRSA. Methods Mol. Biol. 2014, 1085, 131–148. [Google Scholar]
- Rolo, J.; Worning, P.; Nielsen, J.B.; Bowden, R.; Bouchami, O.; Damborg, P.; Guardabassi, L.; Perreten, V.; Tomasz, A.; Westh, H.; et al. Evolutionary Origin of the Staphylococcal Cassette Chromosome mec (SCCmec). Antimicrob. Agents Chemother. 2017, 61, 1042–1046. [Google Scholar] [CrossRef]
- Baig, S.; Johannesen, T.B.; Overballe-Petersen, S.; Larsen, J.; Larsen, A.R.; Stegger, M. Novel SCCmec type XIII (9A) identified in an ST152 methicillin-resistant Staphylococcus aureus. Infect. Genet. Evol. 2018, 61, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Next-generation sequencing to monitor the spread of antimicrobial resistance. Genome. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. [Google Scholar] [CrossRef]
- Otto, M. MRSA virulence and spread. Cell. Microbiol. 2012, 14, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.; Temper, V.; Block, C.S.; Abramson, N.; Moses, A.E. Panton-Valentine Leukocidin–producing Staphylococcus aureus. Emerg. Infect. Dis. 2006, 12, 1789–1790. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, D.R.; Cavaco, L.M.; Nath, G.; Kumar, K.; Gaur, A.; Gokhale, S.; Bhatta, D.R. Association of Panton Valentine Leukocidin (PVL) genes with methicillin resistant Staphylococcus aureus (MRSA) in Western Nepal: A matter of concern for community infections (a hospital based prospective study). BMC Infect. Dis. 2016, 16. [Google Scholar] [CrossRef]
- Özekinci, T.; Dal, T.; Yanık, K.; Özcan, N.; Can, Ş.; Tekin, A.; Yıldırım, H.İ.; Kandemir, İ. Panton-Valentine leukocidin in community and hospital-acquired Staphylococcus aureus strains. Biotechnol. Biotechnol. Equip. 2014, 28, 1089–1094. [Google Scholar] [CrossRef][Green Version]
- Ballhausen, B.; Kriegeskorte, A.; Schleimer, N.; Peters, G.; Becker, K. The mecA Homolog mecC Confers Resistance against β-Lactams in Staphylococcus aureus Irrespective of the Genetic Strain Background. Antimicrob. Agents Chemother. 2014, 58, 3791–3798. [Google Scholar] [CrossRef]
- Paterson, G.K.; Harrison, E.M.; Holmes, M.A. The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2014, 22, 42–47. [Google Scholar] [CrossRef]
- Ariza-Miguel, J.; Hernández, M.; Fernández-Natal, I.; Rodríguez-Lázaro, D. Methicillin-Resistant Staphylococcus aureus Harboring mecC in Livestock in Spain. J. Clin. Microbiol. 2014, 52, 4067–4069. [Google Scholar] [CrossRef]
- Saunders, N.A.; Holmes, A. Multilocus sequence typing (MLST) of Staphylococcus aureus. Methods Mol. Biol. 2007, 391, 71–85. [Google Scholar]
- He, Y.; Xie, Y.; Reed, S. Pulsed-field gel electrophoresis typing of Staphylococcus aureus isolates. Methods Mol. Biol. 2014, 1085, 103–111. [Google Scholar]
- Bosch, T.; de Neeling, A.J.; Schouls, L.M.; Van der Zwaluw, K.W.; Kluytmans, J.A.; Grundmann, H.; Huijsdens, X.W. PFGE diversity within the methicillin-resistant Staphylococcus aureus clonal lineage ST398. BMC Microbiol. 2010, 10, 40. [Google Scholar] [CrossRef]
- Kwon, S.S.; Hong, S.K.; Kim, M.S.; Yong, D.; Lee, K. Performance of Matrix-Assisted Laser Desorption Ionization Time-of-Fight Mass Spectrometry for Rapid Discrimination of Methicillin-Resistant Staphylococcus aureus (MRSA): First Report of a Relation Between Protein Peaks and MRSA spa Type. Ann. Lab. Med. 2017, 37, 553–555. [Google Scholar] [CrossRef]
- Koreen, L.; Ramaswamy, S.V.; Graviss, E.A.; Naidich, S.; Musser, J.M.; Kreiswirth, B.N. spa Typing Method for Discriminating among Staphylococcus aureus Isolates: Implications for Use of a Single Marker to Detect Genetic Micro- and Macrovariation. J. Clin. Microbiol. 2004, 42, 792–799. [Google Scholar] [CrossRef]
- Hallin, M.; Friedrich, A.W.; Struelens, M.J. spa typing for epidemiological surveillance of Staphylococcus aureus. Methods Mol. Biol. 2009, 551, 189–202. [Google Scholar]
- Croxatto, A.; Prod’hom, G.; Greub, G. Applications of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. FEMS Microbiol. Rev. 2012, 36, 380–407. [Google Scholar] [CrossRef]
- Manukumar, H.M.; Umesha, S. MALDI-TOF-MS based identification and molecular characterization of food associated methicillin-resistant Staphylococcus aureus. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Cunningham, S.A.; Chia, N.; Jeraldo, P.R.; Quest, D.J.; Johnson, J.A.; Boxrud, D.J.; Taylor, A.J.; Chen, J.; Jenkins, G.D.; Drucker, T.M.; et al. Comparison of Whole-Genome Sequencing Methods for Analysis of Three Methicillin-Resistant Staphylococcus aureus Outbreaks. J. Clin. Microbiol. 2017, 55, 1946–1953. [Google Scholar] [CrossRef]
- Bhambri, S.; Kim, G. Use of Oral Doxycycline for Community-acquired Methicillin-resistant Staphylococcus aureus (CA-MRSA) Infections. J. Clin. Aesthet. Dermatol. 2009, 2, 45–50. [Google Scholar]
- Cadena, J.; Nair, S.; Henao-Martinez, A.F.; Jorgensen, J.H.; Patterson, J.E.; Sreeramoju, P.V. Dose of Trimethoprim-Sulfamethoxazole To Treat Skin and Skin Structure Infections Caused by Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 5430–5432. [Google Scholar] [CrossRef]
- Archer, G.L.; Coughter, J.P.; Johnston, J.L. Plasmid-encoded trimethoprim resistance in staphylococci. Antimicrob. Agents Chemother. 1986, 29, 733–740. [Google Scholar] [CrossRef]
- Tennent, J.M.; Young, H.K.; Lyon, B.R.; Amyes, S.G.; Skurray, R.A. Trimethoprim resistance determinants encoding a dihydrofolate reductase in clinical isolates of Staphylococcus aureus and coagulase-negative staphylococci. J. Med. Microbiol. 1988, 26, 67–73. [Google Scholar] [CrossRef]
- Larsen, T. Occurrence of doxycycline resistant bacteria in the oral cavity after local administration of doxycycline in patients with periodontal disease. Scand. J. Infect. Dis. 1991, 23, 89–95. [Google Scholar] [CrossRef]
- Handzlik, J.; Matys, A.; Kieć-Kononowicz, K. Recent Advances in Multi-Drug Resistance (MDR) Efflux Pump Inhibitors of Gram-Positive Bacteria S. aureus. Antibiotics (Basel) 2013, 2, 28–45. [Google Scholar] [CrossRef]
- Felicetti, T.; Cannalire, R.; Burali, M.S.; Massari, S.; Manfroni, G.; Barreca, M.L.; Tabarrini, O.; Schindler, B.D.; Sabatini, S.; Kaatz, G.W.; et al. Searching for Novel Inhibitors of the S. aureus NorA Efflux Pump: Synthesis and Biological Evaluation of the 3-Phenyl-1,4-benzothiazine Analogues. ChemMedChem. 2017, 12, 1293–1302. [Google Scholar] [CrossRef]
- Truong-Bolduc, Q.C.; Bolduc, G.R.; Okumura, R.; Celino, B.; Bevis, J.; Liao, C.H.; Hooper, D.C. Implication of the NorB Efflux Pump in the Adaptation of Staphylococcus aureus to Growth at Acid pH and in Resistance to Moxifloxacin. Antimicrob. Agents Chemother. 2011, 55, 3214–3219. [Google Scholar] [CrossRef]
- Spengler, G.; Kincses, A.; Gajdacs, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Klinker, K.P.; Borgert, S.J. Beyond Vancomycin: The Tail of the Lipoglycopeptides. Clin. Ther. 2015, 37, 2619–2636. [Google Scholar] [CrossRef]
- Brunton, L.; Chabner, B.A.; Knollman, B. Goodman & Gillman’s The Pharmacological Basis of Therapeutics, 12th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Moore, C.L.; Lu, M.; Cheema, F.; Osaki-Kiyan, P.; Perri, M.B.; Donabedian, S.; Haque, N.Z.; Zervos, M.J. Prediction of Failure in Vancomycin-Treated Methicillin-Resistant Staphylococcus aureus Bloodstream Infection: A Clinically Useful Risk Stratification Tool. Antimicrob. Agents Chemother. 2011, 55, 4581–4588. [Google Scholar] [CrossRef]
- Bruniera, F.R.; Ferreira, F.M.; Saviolli, L.R.; Bacci, M.R.; Feder, D.; da Luz Gonçalves Pedreira, M.; Sorgini Peterlini, M.A.; Azzalis, L.A.; Campos Junqueira, V.B.; et al. The use of vancomycin with its therapeutic and adverse effects: A review. Eur. Rev. 2015, 19, 694–700. [Google Scholar]
- Gardete, S.; Tomasz, A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J. Clin. Invest. 2014, 124, 2836–2840. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281. [Google Scholar] [PubMed]
- Weinstein, R.A.; Fridkin, S.K. Vancomycin-Intermediate and -Resistant Staphylococcus aureus: What the Infectious Disease Specialist Needs to Know. Clin. Infect. Dis. 2001, 32, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.E.; LeTourneau, D.L.; Hobbs, J.N. Molecular interactions of a semisynthetic glycopeptide antibiotic with D-alanyl-D-alanine and D-alanyl-D-lactate residues. Antimicrob. Agents Chemother. 1997, 41, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chambers, H.F. Staphylococcus aureus with Heterogeneous Resistance to Vancomycin: Epidemiology, Clinical Significance, and Critical Assessment of Diagnostic Methods. Antimicrob. Agents Chemother. 2003, 47, 3040–3045. [Google Scholar] [CrossRef]
- Szabó, J. hVISA/VISA: Diagnostic and therapeutic problems. Expert Rev. Anti-Infect. Ther. 2009, 7, 1–3. [Google Scholar] [CrossRef]
- Guskey, M.T.; Tsuji, B.T. A comparative review of the lipoglycopeptides: Oritavancin, dalbavancin, and telavancin. Pharmacotherapy 2010, 30, 80–94. [Google Scholar] [CrossRef]
- Allen, N.E.; Nicas, T.I. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol. Rev. 2003, 26, 511–532. [Google Scholar] [CrossRef]
- Biedenbach, D.J.; Arhin, F.F.; Moeck, G.; Lynch, T.F.; Sahm, D.F. In vitro activity of oritavancin and comparator agents against staphylococci, streptococci and enterococci from clinical infections in Europe and North America, 2011–2014. Int. J. Antimicrob. Agents 2015, 46, 674–681. [Google Scholar] [CrossRef]
- Leach, K.L.; Swaney, S.M.; Colca, J.R.; McDonald, W.G.; Blinn, J.R.; Thomasco, L.M.; Gadwood, R.C.; Shinabarger, D.; Xiong, L.; Mankin, A.S. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol. Cell 2007, 26, 393–402. [Google Scholar] [CrossRef]
- Falagas, M.E.; Vardakas, K.Z. Benefit-risk assessment of linezolid for serious gram-positive bacterial infections. Drug Saf. 2008, 31, 753–768. [Google Scholar] [CrossRef]
- Long, K.S.; Vester, B. Resistance to Linezolid Caused by Modifications at Its Binding Site on the Ribosome. Antimicrob. Agents Chemother. 2012, 56, 603–612. [Google Scholar] [CrossRef]
- Stefani, S.; Bongiorno, D.; Mongelli, G.; Campanile, F. Linezolid Resistance in Staphylococci. Pharmaceuticals (Basel) 2010, 3, 1988–2006. [Google Scholar] [CrossRef]
- Steenbergen, J.N.; Alder, J.; Thorne, G.M.; Tally, F.P. Daptomycin: A lipopeptide antibiotic for the treatment of serious Gram-positive infections. J. Antimicrob. Chemother. 2005, 55, 283–288. [Google Scholar] [CrossRef]
- Mensa, B.; Howell, G.L.; Scott, R.; DeGrado, W.F. Comparative Mechanistic Studies of Brilacidin, Daptomycin, and the Antimicrobial Peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [Google Scholar] [CrossRef]
- Lalani, T.; Boucher, H.W.; Cosgrove, S.E.; Fowler, V.G.; Kanafani, Z.A.; Vigliani, G.A.; Campion, M.; Abrutyn, E.; Levine, D.P.; Price, C.S.; et al. Outcomes with daptomycin versus standard therapy for osteoarticular infections associated with Staphylococcus aureus bacteraemia. J. Antimicrob. Chemother. 2008, 61, 177–182. [Google Scholar] [CrossRef]
- Tran, T.T.; Munita, J.M.; Arias, C.A. Mechanisms of drug resistance: Daptomycin resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 32–53. [Google Scholar] [CrossRef]
- Anderson, S.D.; Gums, J.G. Ceftobiprole: An extended-spectrum anti-methicillin-resistant Staphylococcus aureus cephalosporin. Ann. Pharmacother. 2008, 42, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Kisgen, J.; Whitney, D. Ceftobiprole, a Broad-Spectrum Cephalosporin with Activity against Methicillin-Resistant Staphylococcus aureus (MRSA). P.T. 2008, 33, 631–641. [Google Scholar]
- Duplessis, C.; Crum-Cianflone, N.F. Ceftaroline: A New Cephalosporin with Activity against Methicillin-Resistant Staphylococcus aureus (MRSA). Clin. Med. Rev. Ther. 2011, 3. [Google Scholar] [CrossRef]
- Farrell, D.J.; Flamm, R.K.; Sader, H.S.; Jones, R.N. Spectrum and potency of ceftaroline tested against leading pathogens causing skin and soft-tissue infections in Europe (2010). Int. J. Antimicrob. Agents 2013, 41, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; James, D.; Kearns, A.; Woodford, N. Pathogens of skin and skin-structure infections in the UK and their susceptibility to antibiotics, including ceftaroline. J. Antimicrob. Chemother. 2015, 70, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, T.R.; Sader, H.S.; Jones, R.N. Antimicrobial activity of ceftobiprole, a novel anti–methicillin-resistant Staphylococcus aureus cephalosporin, tested against contemporary pathogens: Results from the SENTRY Antimicrobial Surveillance Program (2005–2006). Diagn. Microbiol. Infect. Dis. 2008, 61, 86–95. [Google Scholar] [CrossRef]
- Bérenger, R.; Bourdon, N.; Auzou, M.; Leclercq, R.; Cattoir, V. In vitro activity of new antimicrobial agents against glycopeptide-resistant Enterococcus faecium clinical isolates from France between 2006 and 2008. Med. Mal. Infect. 2011, 41, 405–409. [Google Scholar] [CrossRef]
- Urbán, E.; Stone, G.G. Impact of EUCAST ceftaroline breakpoint change on the susceptibility of methicillin-resistant Staphylococcus aureus isolates collected from patients with complicated skin and soft tissue infections. Clin. Microbiol. Infect. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.C.; Basuino, L.; Diep, B.; Hamilton, S.; Chatterjee, S.S.; Chambers, H.F. Ceftobiprole- and Ceftaroline-Resistant Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 2960–2963. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Nelson, J.; Paterson, D.L.; Fowler, V.G.; Howden, B.P.; Cheng, A.C.; Chatfield, M.; Lipman, J.; Van Hal, S.; O’Sullivan, M.; et al. CAMERA2—combination antibiotic therapy for methicillin-resistant Staphylococcus aureus infection: Study protocol for a randomised controlled trial. Trials 2016, 17. [Google Scholar] [CrossRef]
- Livermore, D.M. Tigecycline: What is it, and where should it be used? J. Antimicrob. Chemother. 2005, 56, 611–614. [Google Scholar] [CrossRef]
- Florescu, I.; Beuran, M.; Dimov, R.; Razbadauskas, A.; Bochan, M.; Fichev, G.; Dukart, G.; Babinchak, T.; Cooper, C.A.; Ellis-Grosse, E.J.; et al. Efficacy and safety of tigecycline compared with vancomycin or linezolid for treatment of serious infections with methicillin-resistant Staphylococcus aureus or vancomycin-resistant enterococci: A Phase 3, multicentre, double-blind, randomized study. J. Antimicrob. Chemother. 2008, 62. [Google Scholar] [CrossRef]
- Koomanachai, P.; Crandon, J.L.; Banevicius, M.A.; Peng, L.; Nicolau, D.P. Pharmacodynamic Profile of Tigecycline against Methicillin-Resistant Staphylococcus aureus in an Experimental Pneumonia Model. Antimicrob. Agents Chemother. 2009, 53, 5060–5063. [Google Scholar] [CrossRef]
- Dixit, D.; Madduri, R.P.; Sharma, R. The role of tigecycline in the treatment of infections in light of the new black box warning. Expert. Rev. Anti-Infect. Ther. 2014, 12, 397–400. [Google Scholar] [CrossRef]
- Solomkin, J.S.; Gardovskis, J.; Lawrence, K.; Montravers, P.; Sway, A.; Evans, D.; Tsai, L. IGNITE4: Results of a Phase 3, Randomized, Multicenter, Prospective Trial of Eravacycline vs. Meropenem in the Treatment of Complicated Intra-Abdominal Infections. Clin. Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Sutcliffe, J.A.; O’Brien, W.; Fyfe, C.; Grossman, T.H. Antibacterial Activity of Eravacycline (TP-434), a Novel Fluorocycline, against Hospital and Community Pathogens. Antimicrob. Agents Chemother. 2013, 57, 5548–5558. [Google Scholar] [CrossRef]
- Dougherty, J.A.; Sucher, A.J.; Chahine, E.B.; Shihadeh, K.C. Omadacycline: A New Tetracycline Antibiotic. Ann. Pharmacother. 2018. [Google Scholar] [CrossRef]
- Villano, S.; Steenbergen, J.; Loh, E. Omadacycline: Development of a novel aminomethylcycline antibiotic for treating drug-resistant bacterial infections. Future Microbiol. 2016, 11, 1421–1434. [Google Scholar] [CrossRef]
- Florindo, C.; Costa, A.; Matos, C.; Nunes, S.L.; Matias, A.N.; Duarte, C.M.M.; Rebelo, L.P.N.; Branco, L.C.; Marrucho, I.M. Novel organic salts based on fluoroquinolone drugs: Synthesis, bioavailability and toxicological profiles. Int. J. Pharm. 2014, 469, 179–189. [Google Scholar] [CrossRef]
- Candel, F.J.; Peñuelas, M. Delafloxacin: Design, development and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 881–891. [Google Scholar] [CrossRef]
- McCurdy, S.; Lawrence, L.; Quintas, M.; Woosley, L.; Flamm, R.; Tseng, C.; Cammarata, S. In Vitro Activity of Delafloxacin and Microbiological Response against Fluoroquinolone-Susceptible and Nonsusceptible Staphylococcus aureus Isolates from Two Phase 3 Studies of Acute Bacterial Skin and Skin Structure Infections. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Flamm, R.K.; Rhomberg, P.R.; Huband, M.D.; Farrell, D.J. In Vitro Activity of Delafloxacin Tested against Isolates of Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis. Antimicrob. Agents Chemother. 2016, 60, 6381–6385. [Google Scholar] [CrossRef]
- Park, H.-S.; Kim, H.-J.; Seol, M.J.; Choi, D.R.; Choi, E.C.; Kwak, J.H. In Vitro and In Vivo Antibacterial Activities of DW-224a, a New Fluoronaphthyridone. Antimicrob. Agents Chemother. 2006, 50, 2261–2264. [Google Scholar] [CrossRef] [PubMed]
- Lauderdale, T.L.; Shiau, Y.R.; Lai, J.F.; Chen, H.C.; King, C.H.R. Comparative In Vitro Activities of Nemonoxacin (TG-873870), a Novel Nonfluorinated Quinolone, and Other Quinolones against Clinical Isolates. Antimicrob. Agents Chemother. 2010, 54, 1338–1342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Park, H.S.; Oh, S.H.; Kim, H.S.; Choi, D.R.; Kwak, J.H. Antimicrobial Activity of Zabofloxacin against Clinically Isolated Streptococcus pneumoniae. Molecules 2016, 21, 1562. [Google Scholar] [CrossRef] [PubMed]
- Daneman, N.; Lu, H.; Redelmeier, D.A. Fluoroquinolones and collagen associated severe adverse events: a longitudinal cohort study. BMJ Open 2015, 5, e010077. [Google Scholar] [CrossRef] [PubMed]
- Laue, H.; Weiss, L.; Bernardi, A.; Hawser, S.; Lociuro, S.; Islam, K. In vitro activity of the novel diaminopyrimidine, iclaprim, in combination with folate inhibitors and other antimicrobials with different mechanisms of action. J. Antimicrob. Chemother. 2007, 60, 1391–1394. [Google Scholar] [CrossRef] [PubMed]
- Sincak, C.A.; Schmidt, J.M. Iclaprim, A novel diaminopyrimidine for the treatment of resistant gram-positive infections. Ann. Pharmacother. 2009, 43, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.A.; Hatfield, S.M.; Leyden, J.J. Mupirocin: A topical antibiotic with a unique structure and mechanism of action. Clin. Pharm. 1987, 6, 761–770. [Google Scholar] [PubMed]
- Poovelikunnel, T.; Gethin, G.; Humphreys, H. Mupirocin resistance: Clinical implications and potential alternatives for the eradication of MRSA. J. Antimicrob. Chemother. 2015, 70, 2681–2692. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Hung, W.C.; Tseng, S.P.; Tsai, J.C.; Hsueh, P.R.; Teng, L.J. Fusidic Acid Resistance Determinants in Staphylococcus aureus Clinical Isolates. Antimicrob. Agents Chemother. 2010, 54, 4985–4991. [Google Scholar] [CrossRef]
- Dobie, D.; Gray, J. Fusidic acid resistance in Staphylococcus aureus. Arch. Dis. Child. 2004, 89, 74–77. [Google Scholar] [CrossRef]
- Paukner, S.; Riedl, R. Pleuromutilins: Potent Drugs for Resistant Bugs-Mode of Action and Resistance. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Jacobs, M.R. Retapamulin: A semisynthetic pleuromutilin compound for topical treatment of skin infections in adults and children. Future Microbiol. 2007, 2, 591–600. [Google Scholar] [CrossRef]
- Paukner, S.; Sader, H.S.; Ivezic-Schoenfeld, Z.; Jones, R.N. Antimicrobial Activity of the Pleuromutilin Antibiotic BC-3781 against Bacterial Pathogens Isolated in the SENTRY Antimicrobial Surveillance Program in 2010. Antimicrob. Agents Chemother. 2013, 57, 4489–4495. [Google Scholar] [CrossRef]
- Sader, H.S.; Paukner, S.; Ivezic-Schoenfeld, Z.; Biedenbach, D.J.; Schmitz, F.J.; Jones, R.N. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 2012, 67, 1170–1175. [Google Scholar] [CrossRef]
- Sai, N.; Laurent, C.; Strale, H.; Denis, O.; Byl, B. Efficacy of the decolonization of methicillin-resistant Staphylococcus aureus carriers in clinical practice. Antimicrob. Resist. Infect. Control 2015, 4, 56. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8. [Google Scholar] [CrossRef]
- Scott, R.W.; Tew, G.N. Mimics of Host Defense Proteins; Strategies for Translation to Therapeutic Applications. Curr. Top. Med. Chem. 2017, 17, 576–589. [Google Scholar] [CrossRef]
- Even, S.; Charlier, C.; Nouaille, S.; Ben Zakour, N.L.; Cretenet, M.; Cousin, F.J.; Gautier, M.; Cocaign-Bousquet, M.; Loubière, P.; Le Loir, Y. Staphylococcus aureus virulence expression is impaired by Lactococcus lactis in mixed cultures. Appl. Environ. Microbiol. 2009, 75, 4459–4472. [Google Scholar] [CrossRef]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef]
- Gao, P.; Ho, P.L.; Yan, B.; Sze, K.H.; Davies, J.; Kao, R.Y.T. Suppression of Staphylococcus aureus virulence by a small-molecule compound. Proc. Natl. Acad. Sci. USA 2018, 115, 8003–8008. [Google Scholar] [CrossRef]
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front Pharmacol. 2014, 4. [Google Scholar] [CrossRef]
- Tegos, G.P.; Haynes, M.; Jacob Strouse, J.; Khan, M.M.T.; Bologa, C.G.; Oprea, T.I.; Sklar, L.A. Microbial Efflux Pump Inhibition: Tactics and Strategies. Curr. Pharm. Des. 2011, 17, 1291–1302. [Google Scholar] [CrossRef]
- Wright, G.D. Something old, something new: Revisiting natural products in antibiotic drug discovery. Can. J. Microbiol. 2014, 60, 147–154. [Google Scholar] [CrossRef]
- Brown, D.G.; Lister, T.; May-Dracka, T.L. New natural products as new leads for antibacterial drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 413–418. [Google Scholar] [CrossRef]
- Golkar, Z.; Bagasra, O.; Pace, D.G. Bacteriophage therapy: A potential solution for the antibiotic resistance crisis. J. Infect. Dev. Ctries. 2014, 8, 129–136. [Google Scholar] [CrossRef]
- Love, M.J.; Bhandari, D.; Dobson, R.C.J.; Billington, C. Potential for Bacteriophage Endolysins to Supplement or Replace Antibiotics in Food Production and Clinical Care. Antibiotics (Basel) 2018, 7. [Google Scholar] [CrossRef]
- Clowry, J.; Irvine, A.D.; McLoughlin, R.M. Next-generation anti-Staphylococcus aureus vaccines: A potential new therapeutic option for atopic dermatitis? J. Allergy Clin. Immunol. 2019, 143, 78–81. [Google Scholar] [CrossRef]
- Deresinski, S. Antistaphylococcal vaccines and immunoglobulins: Current status and future prospects. Drugs 2006, 66, 1797–1806. [Google Scholar] [CrossRef]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar] [CrossRef]
- Park, I.Y.; Cho, J.H.; Kim, K.S.; Kim, Y.-B.; Kim, M.S.; Kim, S.C. Helix stability confers salt resistance upon helical antimicrobial peptides. J. Biol. Chem. 2004, 279, 13896–13901. [Google Scholar] [CrossRef]
- Ahn, M.; Jacob, B.; Gunasekaran, P.; Murugan, R.N.; Ryu, E.K.; Lee, G.; Hyun, J.-K.; Cheong, C.; Kim, N.-H.; Shin, S.Y.; et al. Poly-lysine peptidomimetics having potent antimicrobial activity without hemolytic activity. Amino Acids 2014, 46, 2259–2269. [Google Scholar] [CrossRef]
- Cruz, J.; Flórez, J.; Torres, R.; Urquiza, M.; Gutiérrez, J.A.; Guzmán, F.; Ortiz, C.C. Antimicrobial activity of a new synthetic peptide loaded in polylactic acid or poly(lactic-co-glycolic) acid nanoparticles against Pseudomonas aeruginosa, Escherichia coli O157:H7 and methicillin resistant Staphylococcus aureus (MRSA). Nanotechnology 2017, 28, 135102. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, M.; Sahl, H.-G. Defensin-based anti-infective strategies. Int. J. Med. Microbiol. 2014, 304, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef]
- Bergler, H.; Fuchsbichler, S.; Högenauer, G.; Turnowsky, F. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur. J. Biochem. 1996, 242, 689–694. [Google Scholar] [CrossRef]
- Schiebel, J.; Chang, A.; Lu, H.; Baxter, M.V.; Tonge, P.J.; Kisker, C. Staphylococcus aureus FabI: Inhibition, Substrate Recognition and Potential Implications for In Vivo Essentiality. Structure 2012, 20, 802–813. [Google Scholar] [CrossRef]
- Hibbitts, A.; O’Leary, C. Emerging Nanomedicine Therapies to Counter the Rise of Methicillin-Resistant Staphylococcus aureus. Materials (Basel) 2018, 11. [Google Scholar] [CrossRef]
- Knetsch, M.L.W.; Koole, L.H. New Strategies in the Development of Antimicrobial Coatings: The Example of Increasing Usage of Silver and Silver Nanoparticles. Polymers 2011, 3, 340–366. [Google Scholar] [CrossRef]
- Seil, J.T.; Webster, T.J. Antibacterial effect of zinc oxide nanoparticles combined with ultrasound. Nanotechnology 2012, 23, 495101. [Google Scholar] [CrossRef] [PubMed]
- Azam, A.; Ahmed, A.S.; Oves, M.; Khan, M.; Memic, A. Size-dependent antimicrobial properties of CuO nanoparticles against Gram-positive and -negative bacterial strains. Int. J. Nanomedicine 2012, 7, 3527–3535. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Yu, Y.; Chen, Z.; Jin, M.; Yang, D.; Zhao, Z.; Wang, J.; Shen, Z.; Wang, X.; Qian, D.; et al. Nanoalumina promotes the horizontal transfer of multiresistance genes mediated by plasmids across genera. Proc. Natl. Acad. Sci. USA 2012, 109, 4944–4949. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Ohtake, N.; Jona, H.; Shimizu, A.; Moriya, M.; Sato, H.; Sugimoto, Y.; Ikeura, C.; Kiyonaga, H.; Nakano, M.; et al. Dicationic dithiocarbamate carbapenems with anti-MRSA activity. Bioorg. Med. Chem. 2001, 9, 1571–1578. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Warner, M. Activity of the anti-MRSA carbapenem razupenem (PTZ601) against Enterobacteriaceae with defined resistance mechanisms. J. Antimicrob. Chemother. 2009, 64, 330–335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sunagawa, M.; Itoh, M.; Kubota, K.; Sasaki, A.; Ueda, Y.; Angehrn, P.; Bourson, A.; Goetschi, E.; Hebeisen, P.; Then, R.L. New anti-MRSA and anti-VRE carbapenems; synthesis and structure-activity relationships of 1beta-methyl-2-(thiazol-2-ylthio)carbapenems. J. Antibiot. 2002, 55, 722–757. [Google Scholar] [CrossRef][Green Version]
- Zhou, M.; Chen, J.; Liu, Y.; Hu, Y.; Liu, Y.; Lu, J.; Zhang, S.; Yu, Y.; Huang, X.; Yang, Q.; et al. In Vitro Activities of Ceftaroline/Avibactam, Ceftazidime/Avibactam, and Other Comparators Against Pathogens From Various Complicated Infections in China. Clin. Infect. Dis. 2018, 67, S206–S216. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Johnstone, M.R.; Ross, P.L.; McLaughlin, R.E.; Olivier, N.B.; Alm, R.A. Avibactam and Class C β-Lactamases: Mechanism of Inhibition, Conservation of the Binding Pocket, and Implications for Resistance. Antimicrob. Agents Chemother. 2014, 58, 5704–5713. [Google Scholar] [CrossRef]
- Luo, M.L.; Leenay, R.T.; Beisel, C.L. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol. Bioeng. 2016, 113, 930–943. [Google Scholar] [CrossRef]
- Yanagihara, K.; Tashiro, M.; Fukuda, Y.; Ohno, H.; Higashiyama, Y.; Miyazaki, Y.; Hirakata, Y.; Tomono, K.; Mizuta, Y.; Tsukamoto, K.; et al. Effects of short interfering RNA against methicillin-resistant Staphylococcus aureus coagulase in vitro and in vivo. J. Antimicrob. Chemother. 2006, 57, 122–126. [Google Scholar] [CrossRef]
- Spellberg, B. The future of antibiotics. Crit. Care 2014, 18, 228. [Google Scholar] [CrossRef]
- Gajdács, M.; Handzlik, J.; Sanmartín, C.; Domínguez-Álvarez, E.; Spengler, G. Prediction of ADME properties for selenocompounds with anticancer and efflux pump inhibitory activity using preliminary computational methods (article in Hungarian). Acta Pharm. Hung. 2018, 88, 67–74. [Google Scholar]
- Aung, A.K.; Haas, D.W.; Hulgan, T.; Phillips, E.J. Pharmacogenomics of antimicrobial agents. Pharmacogenomics 2014, 15, 1903–1930. [Google Scholar] [CrossRef]
- Weng, Z.; DeLisi, C. Protein therapeutics: Promises and challenges for the 21st century. Trends Biotechnol. 2002, 20, 29–35. [Google Scholar] [CrossRef]
- Ruppé, E.; Burdet, C.; Grall, N.; de Lastours, V.; Lescure, F.-X.; Andremont, A.; Armand-Lefèvre, L. Impact of antibiotics on the intestinal microbiota needs to be re-defined to optimize antibiotic usage. Clin. Microbiol. Infect. 2018, 24, 3–5. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Antibiotic Class (with Examples) | Advantages Indications (in Italics) | Disadvantages |
|---|---|---|
| SMX/TMP | Available for oral and parenteral use Good tolerability Price of therapy Wide range of indications | Resistance levels iv. infusion has to be administered in a large volume of fluid |
| Tetracyclines/Glycylcyclines (doxycycline, tygecycline) | Broad spectrum activity Wide range of indications (tigecycline: SSTIs, cIAI, CAP) | Doxycycline: resistance levels Tygecycline: black box warning, iv. only Severe nausea and vomiting (dose-limiting side effect) |
| Novel tetracycline-derivatives (eravacycline, omadacycline) | Broad spectrum activity CAP, SSTIs | Severe nausea and vomiting (dose-limiting side effect) Parenteral only Resistance expression/horizontally transmitted resistance genes |
| Glycopeptides (vancomycin, teicoplainin) | Gold standard of MRSA-therapy for a long time Extensive clinical data available regarding its usePrice of therapy Wide range of indications | MIC creep Parenteral only (with exceptions) TDM required (due to nephrotoxicity and ototoxicity) Resistance expression (hVISA, VISA, VRSA) |
| Lipoglycopeptides (telavancin, dalbavancin, oritavancin) | Long half-life (single-dose therapy) Useful in OPAT There is no need for TDM SSTIs, bone and joint infections HAP, VAP (telavancin) | Parenteral only Price of therapy Cannot be removed by dialysis Increased mortality in renal insufficiency Resistance expression/horizontally transmitted resistance genes |
| Oxazolidinones (linezolid, tedizolid) | Available for oral and parenteral use SSTIs, bone and joint infections | Drug-drug interactions MAO-inhibition (Serotonin-syndrome) Price of therapy Resistance expression/horizontally transmitted resistance genes |
| Lipopeptides (daptomycin) | Bloodstream infections, infective endocarditis, SSTIs | Not useful in pneumonia Parenteral only Resistance expression/horizontally transmitted resistance genes |
| 5th generation cephalosporins (ceftaroline, ceftobiprole) | Good tolerability SSTIs, CAP, HAP, MRSA bacteremia | Price of therapy Hydrolized by ESBLs (mixed infections) Resistance expression/horizontally transmitted resistance genes |
| Older fluoroquinolones (ciprofloxacin, levofloxacin, moxifloxacin) | Available for oral and parenteral use Extensive clinical data available regarding their use Good tolerability Accumulation in the intracellular space Price of therapy Broad-spectrum activity Wide range of indications | Side effect profile (especially in light of recent developments) Resistance levels and rapid resistance development |
| Next-generation fluoroquinolones (delafloxacin; avarofloxacin, finafloxacin, zaborfloxacin, nemonoxacin) | Available for oral and parenteral use Broad-spectrum activity Accumulation in the intracellular space Presently studied in a wide range of indications (e.g., cSSTI, CAP, HAP, cUTI MDR gonorrhea) | Black box warining Side effect profile Price of therapy |
| Mupirocin | Price of therapy Dose-dependent bactericidal activity Topical agent for MRSA nasal decolonization Additonal indications are being studied | Resistance development Risk of toxicity when used orally/parenterally |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajdács, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. https://doi.org/10.3390/antibiotics8020052
Gajdács M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics. 2019; 8(2):52. https://doi.org/10.3390/antibiotics8020052
Chicago/Turabian StyleGajdács, Márió. 2019. "The Continuing Threat of Methicillin-Resistant Staphylococcus aureus" Antibiotics 8, no. 2: 52. https://doi.org/10.3390/antibiotics8020052
APA StyleGajdács, M. (2019). The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics, 8(2), 52. https://doi.org/10.3390/antibiotics8020052