Peptidoglycan O-Acetylation as a Virulence Factor: Its Effect on Lysozyme in the Innate Immune System


Abstract
:1. Introduction
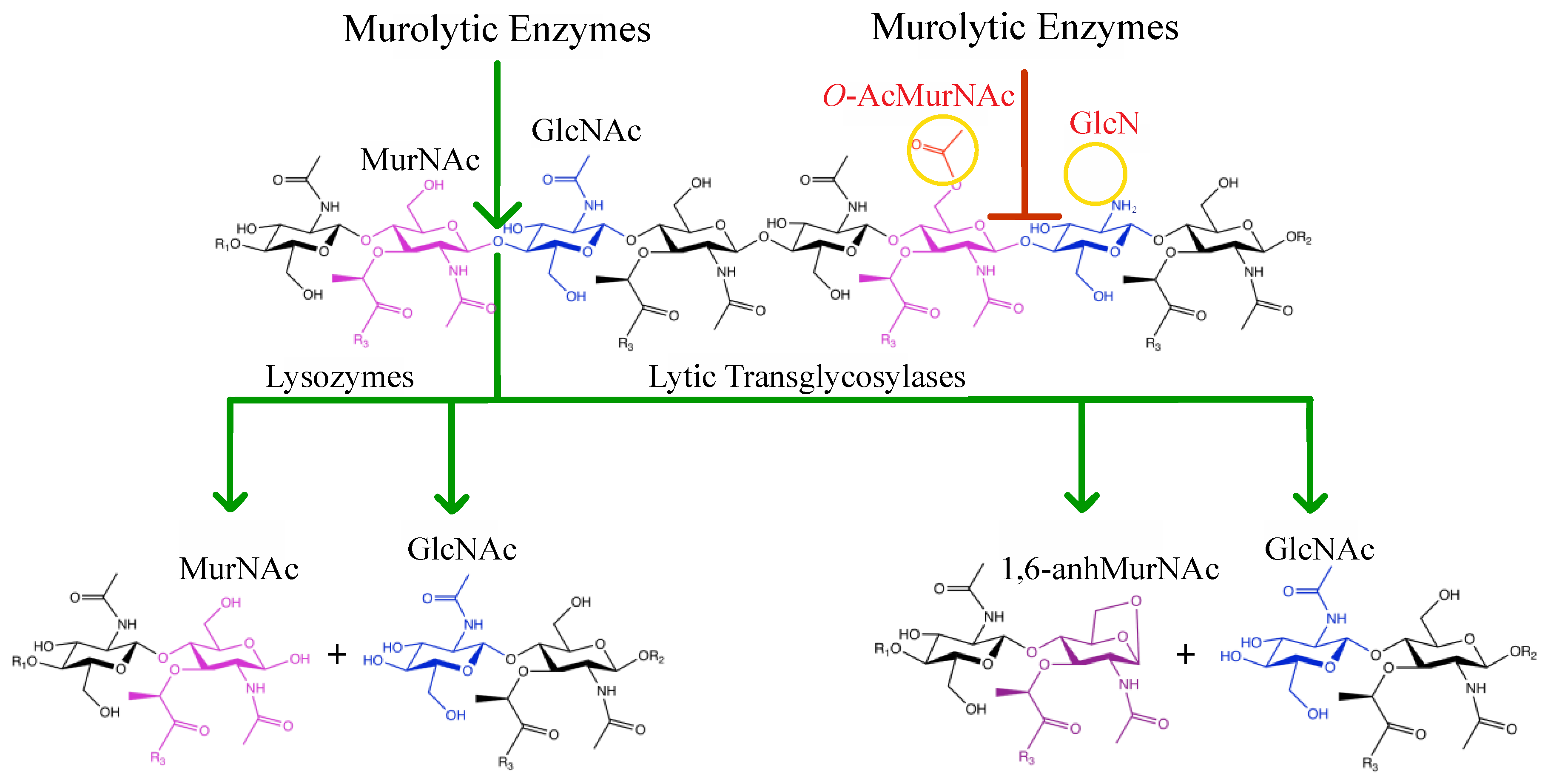
2. Peptidoglycan Composition
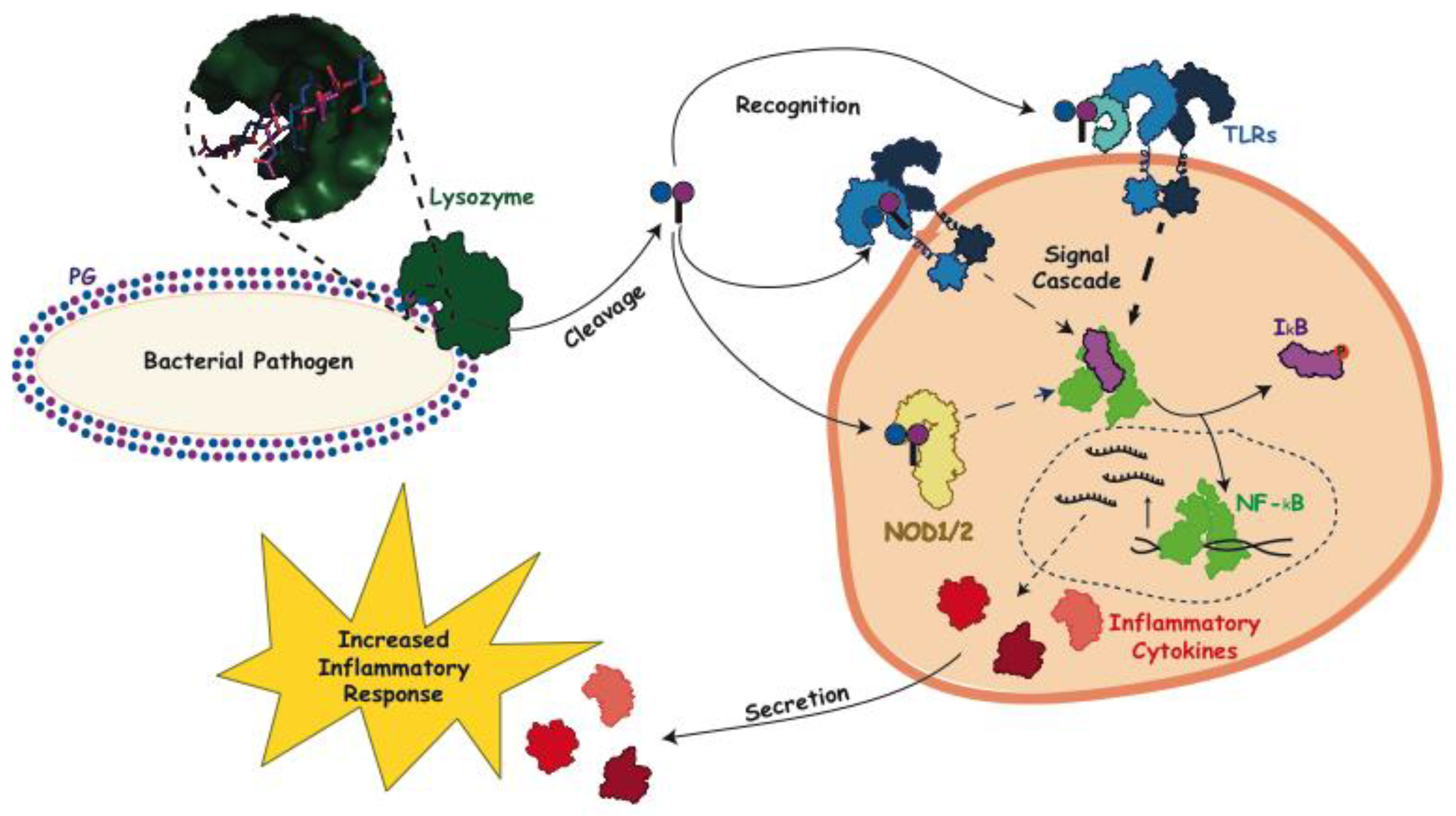
3. Peptidoglycan and Host Immune Interactions
4. Interaction of Lysozyme with PG
5. O-Acetylation of Peptidoglycan
6. Physiological and Pathobiological Significance of PG O-Acetylation
6.1. Physiological Role
6.2. Pathobiology of PG O-Acetylation
7. Discussion: Targeting O-Acetylation as a Novel Anti-Virulence Target
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance. 2014. Available online: https://apps.who.int/iris/bitstream/handle/10665/112642/9789241564748_eng.pdf?sequence=1 (accessed on 5 June 2019).
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. 2018. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 5 June 2019).
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. 2013. Available online: https://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf (accessed on 5 June 2019).
- Brown, E.D.; Wright, W.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Sychantha, D.; Brott, A.S.; Jones, C.S.; Clarke, A.J. Mechanistic pathways for peptidoglycan O-acetylation and de-O-acetylation. Front. Microbiol. 2018, 9, 2332. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef]
- Pushkaran, A.C.; Nataraj, N.; Nair, N.; Götz, F.; Biswas, R.; Mohan, C.G. Understanding the structure-function relationship of lysozyme resistance in Staphylococcus aureus by peptidoglycan O-acetylation using molecular docking, dynamics, and lysis assay. J. Chem. Inf. Model. 2015, 55, 760–770. [Google Scholar] [CrossRef]
- Clarke, A.J.; Strating, H.; Blackburn, N.T. Pathways for the O-acetylation of bacterial cell wall polysaccharides. In Glycomicrobiology; Doyle, R.J., Ed.; Plenum Publishing Co Ltd.: New York, NY, USA, 2002; pp. 187–223. [Google Scholar]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan stricture and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Ward, J.B. The chain length of the glycans in bacterial cell walls. Biochem. J. 1973, 133, 395–398. [Google Scholar] [CrossRef]
- Costa, K.; Bacher, G.; Allmaier, G.; Dominguez-Bello, M.G.; Engstrand, L.; Falk, P.; de Pedro, M.A.; García-del Portillo, F. The morphological transition of Helicobacter pylori cells from spiral to coccoid is preceded by a substantial modification of the cell wall. J. Bacteriol. 1999, 181, 3710–3715. [Google Scholar]
- Glauner, B. Separation and quantification of muropeptides with high-performance liquid chromatography. Anal. Biochem. 1988, 172, 451–464. [Google Scholar] [CrossRef]
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar]
- Siewert, G.; Strominger, J.L. Biosynthesis of the peptidoglycan of bacterial cell walls. XI. Formation of the isoglutamine amide group in the cell walls of Staphylococcus aureus. J. Biol. Chem. 1968, 243, 783–790. [Google Scholar]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef] [Green Version]
- Mirelman, D.; Sharon, N. Isolation and characterization of two disaccharide-peptides from lysozyme digests of Micrococcus lysodeikticus cell walls. Biochem. Biophys. Res. Commun. 1966, 24, 237–243. [Google Scholar] [CrossRef]
- Jutras, B.; Lochhead, R.B.; Kloos, Z.A.; Biboy, J.; Strle, K.; Booth, C.J.; Govers, S.K.; Peter, S.; Waldemar, V.; Linda, K.B.; et al. Borrelia burgdorferi peptidoglycan is a persistent antigen in patients with Lyme arthritis. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Vollmer, W. Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol. Rev. 2008, 32, 287–306. [Google Scholar] [CrossRef]
- Fleming, A. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. B 1922, 93, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Callewart, L.; Michiels, C.W. Lysozymes in the animal kingdom. J. Biosci. 2010, 35, 127–160. [Google Scholar] [CrossRef]
- Derde, M.; Lechevalier, V.; Guérin-Dubiard, C.; Cochet, M.F.; Jan, S.; Baron, F.; Gautier, M.; Vié, V.; Nau, F. Hen egg white lysozyme permeabilizes Escherichia coli outer and inner membranes. J. Agric. Food. Chem. 2013, 61, 9922–9929. [Google Scholar] [CrossRef]
- Salton, M.R.J. Cell wall of Micrococcus lysodeikticus as the substrate of lysozyme. Nature 1952, 170, 746–747. [Google Scholar] [CrossRef]
- Ellison, R.T., III; Giehl, T.J. Killing Gram-negative bacteria by lactoferrin and lysozyme. J. Clin. Invest. 1991, 88, 1080–1091. [Google Scholar] [CrossRef]
- Hancock, R.E.; Scott, M.G. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861. [Google Scholar] [CrossRef] [Green Version]
- Sukhithasri, V.; Nisha, N.; Biswas, L.; Anil Kumar, V.; Biswas, R. Innate immune recognition of microbial cell wall components and microbial strategies to evade such recognitions. Microbiol. Res. 2013, 168, 396–406. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Philpott, D.J. Peptidoglycan: A critical activator of the mammalian immune system during infection and homeostasis. Immunol. Rev. 2011, 243, 40–60. [Google Scholar] [CrossRef]
- Davis, K.M.; Nakamura, S.; Weiser, J.N. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J. Clin. Invest. 2011, 121, 3666–3676. [Google Scholar] [CrossRef]
- Takeuchi, O.; Hoshino, K.; Kawai, T.; Sanjo, H.; Takada, H.; Ogawa, T.; Takeda, K.; Akira, S. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity 1999, 11, 443–451. [Google Scholar] [CrossRef]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef]
- Schaefer, A.K.; Meinyk, J.E.; Baksh, M.M.; Lazor, K.M.; Finn, M.G.; Grimes, C.L. Membrane association dictates ligand specificity for the innate immune receptor NOD2. ACS Chem. Biol. 2017, 12, 2216–2224. [Google Scholar] [CrossRef]
- Knilans, K.J.; Hackett, K.T.; Anderson, J.E.; Weng, C.; Dillard, J.P.; Duncan, J.A. Neisseria gonorrhoeae lytic transglycosylases LtgA and LtgD reduce host innate immune signaling through TLR2 and NOD2. ACS Infect. Dis. 2017, 3, 624–633. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Was, M.N.; Sternberd, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Blake, C.C.; Koenig, D.F.; Mair, G.A.; North, A.C.; Philips, D.C.; Sarma, V.R. Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Å resolution. Nature 1965, 206, 757–761. [Google Scholar] [CrossRef]
- Vocadlo, D.J.; Davies, G.J.; Laine, R.; Withers, S.G. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.C. The hen egg-white lysozyme molecule. Proc. Natl. Acad. Sci. USA 1967, 57, 483–495. [Google Scholar] [CrossRef]
- Brumfitt, W.; Wardlaw, A.C.; Park, J.T. Development of lysozyme-resistance in Micrococcus lysodeikticus and its association with an increased O-acetyl content of the cell wall. Nature 1958, 181, 1783–1784. [Google Scholar] [CrossRef]
- Clarke, A.J. The extent of peptidoglycan O-acetylation in the tribe Proteeae. J. Bacteriol. 1993, 175, 4550–4553. [Google Scholar] [CrossRef]
- Clarke, C.A.; Scheurwater, E.M.; Clarke, A.J. The vertebrate lysozyme inhibitor Ivy functions to inhibit the activity of lytic transglycosylase. J. Biol. Chem. 2010, 285, 14843–14847. [Google Scholar] [CrossRef] [PubMed]
- Sychantha, D.; Little, D.J.; Chapman, R.N.; Boons, G.J.; Robinson, H.; Howell, P.L.; Clarke, A.J. PatB1 is an O-acetyltransferase that decorates secondary cell wall polysaccharides. Nat. Chem. Biol. 2018, 14, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Clarke, A.J. Dependence of lysozyme-catalysed solubilization of Proteus mirabilis peptidoglycan on the extent of O-acetylation. Eur. J. Biochem. 1991, 195, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.S.; Folkening, W.J.; Miller, D.R.; Swim, S.C. Resistance of O-acetylated gonococcal peptidoglycan to human peptidoglycan-degrading enzymes. Infect. Immun. 1983, 40, 903–911. [Google Scholar] [PubMed]
- Swim, S.C.; Gfell, M.A.; Wilde, C.E., III; Rosenthal, R.S. Strain distribution in extents of lysozyme resistance and O-acetylation of gonococcal peptidoglycan determined by high-performance liquid chromatography. Infect. Immun. 1983, 42, 446–452. [Google Scholar] [Green Version]
- Rosenthal, R.S.; Blundell, J.K.; Perkins, H.R. Strain-related differences in lysozyme sensitivity and extent of O-acetylation of gonococcal peptidoglycan. Infect. Immun. 1982, 37, 826–829. [Google Scholar] [PubMed]
- Blundell, J.K.; Smith, G.J.; Perkins, H.R. The peptidoglycan of Neisseria gonorrhoeae: O-acetyl groups and lysozyme sensitivity. FEMS Microbiol. Lett. 1980, 9, 259–261. [Google Scholar] [CrossRef]
- Bera, A.; Biswas, R.; Herbert, S.; Götz, F. The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect Immun. 2006, 74, 4598–4604. [Google Scholar] [CrossRef] [PubMed]
- Lear, A.L.; Perkins, H.R. Degrees of O-acetylation and cross-linking of the peptidoglycan of Neisseria gonorrhoeae during growth. J. Gen. Microbiol. 1983, 129, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, J.; Kroll, H.P. Murein biosynthesis and O-acetylation of N-acetylmuramic acid during the cell-division cycle of Proteus mirabilis. Eur. J. Biochem. 1981, 117, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J. Peptidoglycan biosynthesis in Neisseria gonorrhoeae strains sensitive and intrinsically resistant to beta-lactam antibiotics. J. Bacteriol. 1983, 153, 429–435. [Google Scholar] [PubMed]
- Blundell, J.K.; Perkins, H.R. Effects of beta-lactam antibiotics on peptidoglycan synthesis in growing Neisseria gonorrhoeae, including changes in the degree of O-acetylation. J. Bacteriol. 1981, 147, 633–641. [Google Scholar]
- Martin, H.H.; Gmeiner, J. Modification of peptidoglycan structure by penicillin action in the cell walls of Proteus mirabilis. Eur. J. Biochem. 1979, 95, 487–495. [Google Scholar] [CrossRef]
- Sidow, T.; Johannsen, L.; Labischinski, H. Penicillin-induced changes in the cell wall composition in Staphylococcus aureus before the onset of bacteriolysis. Arch. Microbiol. 1990, 154, 73–81. [Google Scholar] [CrossRef]
- Weadge, H.T.; Clarke, A.J. Identification of a new family of enzymes with potential O-acetylpeptidoglycan esterase activity in both Gram-positive and Gram-negative bacteria. BMC Microbiol. 2005, 5, 49. [Google Scholar] [CrossRef]
- Franklin, M.J.; Ohman, D.E. Mutant analysis and cellular localization of the AlgI, AlgJ and AlgF proteins required for O-acetylation of alginate in Pseudomonas aeruginosa. J. Bacteriol. 2002, 184, 3000–3007. [Google Scholar] [CrossRef]
- Dillard, J.P.; Hackett, K.T. Mutations affecting peptidoglycan acetylation in Neisseria gonorrhoeae and Neisseria meningitidis. Infect. Immun. 2005, 73, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, P.J.; Clarke, A.J. O-Acetylation of peptidoglycan in Gram-negative bacteria: Identification and characterization of PG O-acetyltransferase in Neisseria gonorrhoeae. J. Biol. Chem. 2010, 285, 13264–13273. [Google Scholar] [CrossRef] [PubMed]
- Weadge, J.T.; Clarke, A.J. Identification and characterization of O-acetylpeptidoglycan esterase, a novel enzyme discovered in N. gonorrhoeae. Biochemistry 2006, 45, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Götz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Crisóstomo, M.L.; Vollmer, W.; Kharat, A.S.; Inhülsen, S.; Gehre, F.; Buckenmaier, S.; Tomasz, A. Attenuation of penicillin resistance in peptidoglycan O-acetyl transferase mutant of Streptococcus pneumoniae. Mol. Microbiol. 2006, 61, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Sychantha, D.; Jones, S.; Little, D.J.; Moynihan, P.J.; Robinson, H.; Galley, N.F.; Roper, D.I.; Dowson, C.G.; Howell, P.L.; Clarke, A.J. In vitro characterization of the antivirulence target of Gram-positive pathogens, peptidoglycan O-acetyltransferase A (OatA). PLOS Pathog. 2017, 13, e1006667. [Google Scholar] [CrossRef]
- Sychantha, D.; Clarke, A.J. Peptidoglycan modification by the catalytic domain of Streptococcus pneumoniae OatA follows a ping-pong bi-bi mechanism of action. Biochemistry 2018, 57, 2394–2401. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, P.J.; Clarke, A.J. Assay for peptidoglycan O-acetyltransferase: A potential new antibacterial target. Anal. Biochem. 2013, 439, 73–79. [Google Scholar] [CrossRef]
- Moynihan, P.J.; Clarke, A.J. Substrate specificity and kinetic characterization of peptidoglycan O-acetyltransferase B from Neisseria gonorrhoeae. J. Biol. Chem. 2014, 289, 16748–16760. [Google Scholar] [CrossRef]
- Moynihan, P.J.; Clarke, A.J. The mechanism of peptidoglycan O-acetyltransferase involves an Asp-His-Ser catalytic triad. Biochemistry 2014, 53, 6243–6251. [Google Scholar] [CrossRef]
- Scheurwater, E.M.; Reid, C.W.; Clarke, A.J. Lytic transglycosylases: Bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 2008, 40, 586–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höltje, J.-V.; Mirelman, D.; Sharon, N.; Schwarz, U. Novel type of murein transglycosylase in Escherichia coli. J. Bacteriol. 1975, 124, 1067–1076. [Google Scholar]
- Moynihan, P.J.; Clarke, A.J. O-Acetylated peptidoglycan: Controlling the activity of bacterial autolysins and lytic enzymes of the innate immune system. Int. J. Biochem. Cell Biol. 2011, 43, 1655–1659. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, N.T.; Clarke, A.J. Characterization of soluble and membrane-bound family 3 lytic transglycosylase from Pseudomonas aeruginosa. Biochemistry 2002, 41, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Höltje, J.-V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998, 62, 181–203. [Google Scholar]
- Naninga, N. Cell division and peptidoglycan assembly in Escherichia coli. Mol. Microbiol. 1991, 5, 791–795. [Google Scholar] [CrossRef]
- Ghuysen, J.-M. Serine β-lactamases and penicillin-binding proteins. Annu. Rev. Microbiol. 1991, 45, 37–67. [Google Scholar] [CrossRef]
- Bernard, E.; Rolain, T.; David, B.; André, G.; Dupres, V.; Dufrêne, Y.F.; Hallet, B.; Chapot-Chartier, M.P.; Hols, P. Dual role for the O-acetyltransferase OatA in peptidoglycan modification and control of cell septation in Lactobacillus plantarum. PLoS ONE 2012, 7, e47893. [Google Scholar] [CrossRef]
- Seidl, P.H.; Schleifer, K.H. (Eds.) Biological Properties of Peptidoglycan; Walter de Gruyter: New York, NY, USA, 1985. [Google Scholar]
- Clarke, A.J.; Dupont, C. O-Acetylated peptidoglycan: Its occurrence, pathobiological significance and biosynthesis. Can. J. Microbiol. 1991, 38, 85–91. [Google Scholar] [CrossRef]
- Biberstine, K.J.; Darr, D.S.; Rosenthal, R.S. Tolerance to appetite suppression induced by peptidoglycan. Infect. Immun. 1996, 64, 3641–3645. [Google Scholar] [Green Version]
- Biberstine, K.J.; Rosenthal, R.S. Peptidoglycan fragments decrease food intake and body weight gain in rats. Infect. Immun. 1994, 62, 3276–3281. [Google Scholar] [PubMed]
- Baranwal, G.; Mohammad, M.; Jarneborn, A.; Reddy, B.R.; Golla, A.; Chakravarty, S.; Biswas, L.; Götz, F.; Shankarappa, S.; Jin, T.; et al. Impact of cell wall peptidoglycan O-acetylation on the pathogensis of Staphylococcus aureus in septic arthritis. Int. J. Med. Microbiol. 2017, 307, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.J.; Wallsmith, D.E.; Rosenthal, R.S. Arthropathic properties of gonococcal peptidoglycan fragments: Implications for the pathogenesis of disseminated gonococcal disease. Infect. Immun. 1986, 52, 600–608. [Google Scholar] [PubMed]
- Shimada, T.; Park, B.G.; Wolf, A.J.; Brikos, C.; Goodridge, H.S.; Becker, C.A.; Reyes, C.N.; Miao, E.A.; Aderem, A.; Götz, F.; et al. Staphylococcus aureus evades the lysozyme-based digestion of peptidoglycan that links phagocytosis and macrophage IL-β1 secretion. Cell Host Microbe 2010, 7, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.; Kolar, S.L.; Müller, S.; Reyes, C.N.; Wolf, A.J.; Ogawa, C.; Singhania, R.; De Caralho, D.D.; Ardit, M.; Underhill, D.M.; et al. O-Acetylation of peptidoglycan limits helper T cell priming and permits Staphylococcus aureus reinfection. Cell Host Microbe 2017, 22, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.M.; Akinbi, H.T.; Standish, A.J.; Weiser, J.N. Resistance to mucosal lysozyme compensates for the fitness deficit of peptidoglycan modifications by Streptococcus pneumoniae. PLoS Pathog. 2008, 4, e1000241. [Google Scholar] [CrossRef] [PubMed]
- Aubry, C.; Goulard, C.; Nahori, M.A.; Cayet, N.; Decalf, J.; Sachse, M.; Boneca, I.G.; Cossart, P.; Dussurget, O. OatA, a peptidoglycan O-acetyltransferase involved in Listeria monocytogenes immune escape, is critical for virulence. J. Infect. Dis. 2011, 204, 731–740. [Google Scholar] [CrossRef]
- Wang, G.; Lo, L.F.; Forsberg, L.S.; Maier, R.J. Helicobacter pylori peptidoglycan modifications confer lysozyme resistance and contribute to survival in the host. mBio 2012, 3, e00409-12. [Google Scholar] [CrossRef]
- Veyrier, F.J.; Williams, A.H.; Mesnage, S.; Schmitt, C.; Taha, M.K.; Boneca, I.G. De-O-acetylation of peptidoglycan regulates glycan chain extension and affects in vivo survival of Neisseria meningitidis. Mol. Microbiol. 2013, 87, 1100–1112. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef]
- Mühlen, S.; Dersch, P. Anti-virulence strategies to target bacterial infections. In How to Overcome the Antibiotic Crisis. Current Topics in Microbiology and Immunity; Mühlen, S., Dersch, P., Eds.; Springer: Cham, Switzerland, 2015; Volume 398, pp. 147–168. [Google Scholar]
- Brott, A.S.; Jones, C.S.; Clarke, A.J. Development of a high throughput screen for the identification of peptidoglycan O-acetyltransferases, new potential antibacterial targets. Antibiotics 2019, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.P.; Neely, M.N. CpsY influences Streptococcus iniae cell wall adaptations important for neutrophil intracellular survival. Infect. Immun. 2012, 80, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Wichgers Schreur, P.J.; van Weeghel, C.; Rebel, J.M.; Smits, M.A.; van Putten, J.P.; Smith, H.E. Lysozyme resistance in Streptococcus suis is highly variable and multifactorial. PLoS ONE 2012, 7, e36281. [Google Scholar] [CrossRef] [PubMed]
- Hébert, L.; Courtin, P.; Torelli, R.; Sanguinetti, M.; Chapot-Chartier, M.P.; Auffray, Y.; Benachour, A. Enterococcus faecalis constitutes an unusual bacterial model in lysozyme resistance. Infect. Immun. 2007, 75, 5390–5398. [Google Scholar] [CrossRef] [PubMed]
- Le Jeune, A.; Torelli, R.; Sanguinetti, M.; Giard, J.C.; Hartke, A.; Auffray, Y.; Benachour, A. The extracytoplasmic function sigma factor SigV plays a key role in the original model of lysozyme resistance and virulence of Enterococcus faecalis. PLoS ONE 2010, 5, e9658. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.P.; Loukitcheva, A.; Zemansky, J.; Wheeler, R.; Boneca, I.G.; Portnoy, D.A. Listeria monocytogenes is resistant to lysozyme through the regulation, not the acquisition, of cell wall-modifying enzymes. J. Bacteriol. 2014, 196, 3756–3767. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gram-positive | Gram-negative |
|---|---|
| Clostridium difficile | Enterobacteriaceae3 |
| Enterococcus (faecium) | Neisseria gonorrhoeae |
| Staphylococcus aureus (incl. MRSA/VRSA) | Acinetobacter |
| Mycobacterium tuberculosis | Helicobacter pylori |
| Streptococcus pneumoniae | Campylobacterspp. |
| Group A/B Streptococcus | Shigella spp. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brott, A.S.; Clarke, A.J. Peptidoglycan O-Acetylation as a Virulence Factor: Its Effect on Lysozyme in the Innate Immune System. Antibiotics 2019, 8, 94. https://doi.org/10.3390/antibiotics8030094
Brott AS, Clarke AJ. Peptidoglycan O-Acetylation as a Virulence Factor: Its Effect on Lysozyme in the Innate Immune System. Antibiotics. 2019; 8(3):94. https://doi.org/10.3390/antibiotics8030094
Chicago/Turabian StyleBrott, Ashley S., and Anthony J. Clarke. 2019. "Peptidoglycan O-Acetylation as a Virulence Factor: Its Effect on Lysozyme in the Innate Immune System" Antibiotics 8, no. 3: 94. https://doi.org/10.3390/antibiotics8030094
APA StyleBrott, A. S., & Clarke, A. J. (2019). Peptidoglycan O-Acetylation as a Virulence Factor: Its Effect on Lysozyme in the Innate Immune System. Antibiotics, 8(3), 94. https://doi.org/10.3390/antibiotics8030094