Pangenome Analysis of Mycobacterium tuberculosis Reveals Core-Drug Targets and Screening of Promising Lead Compounds for Drug Discovery

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
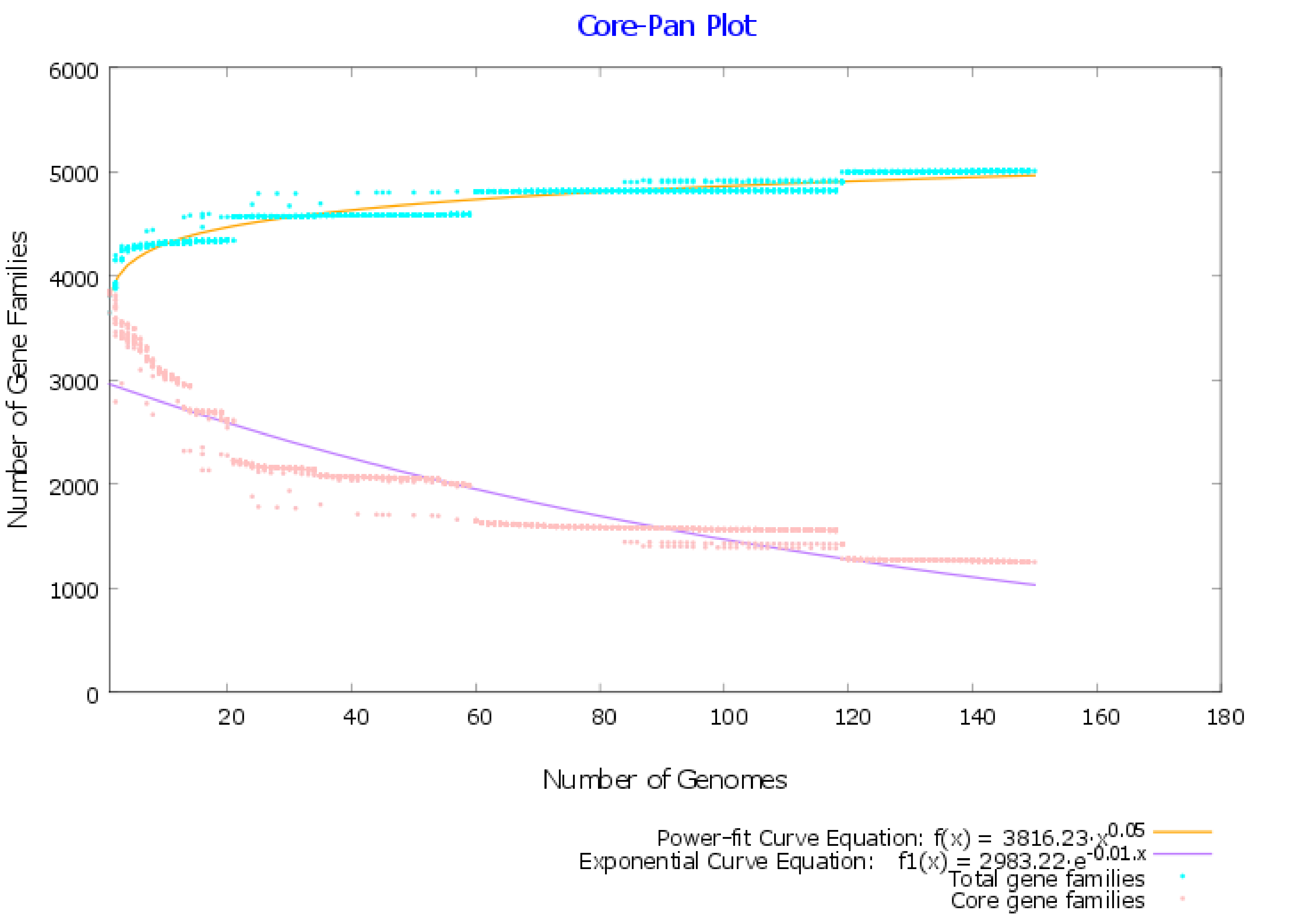
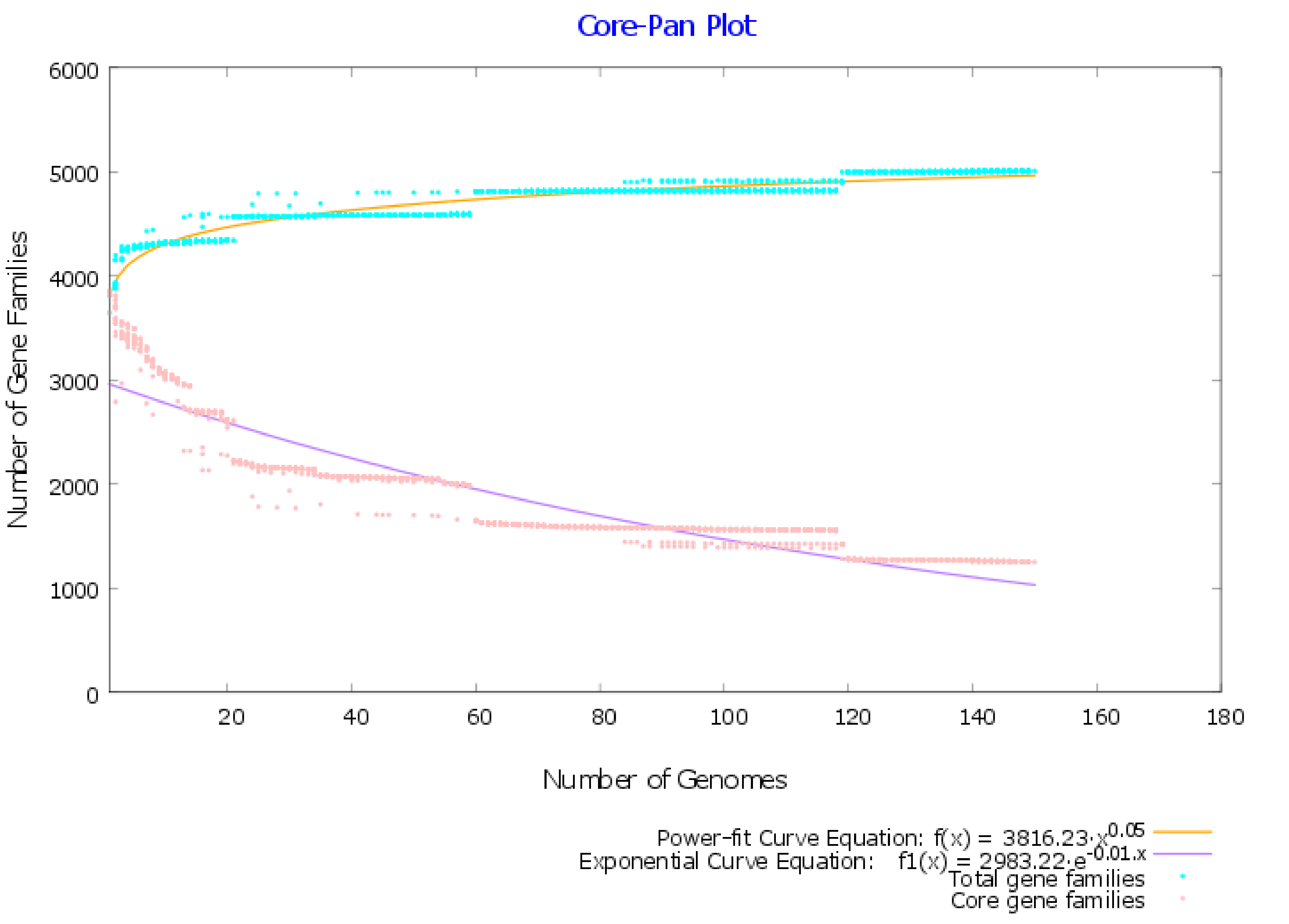
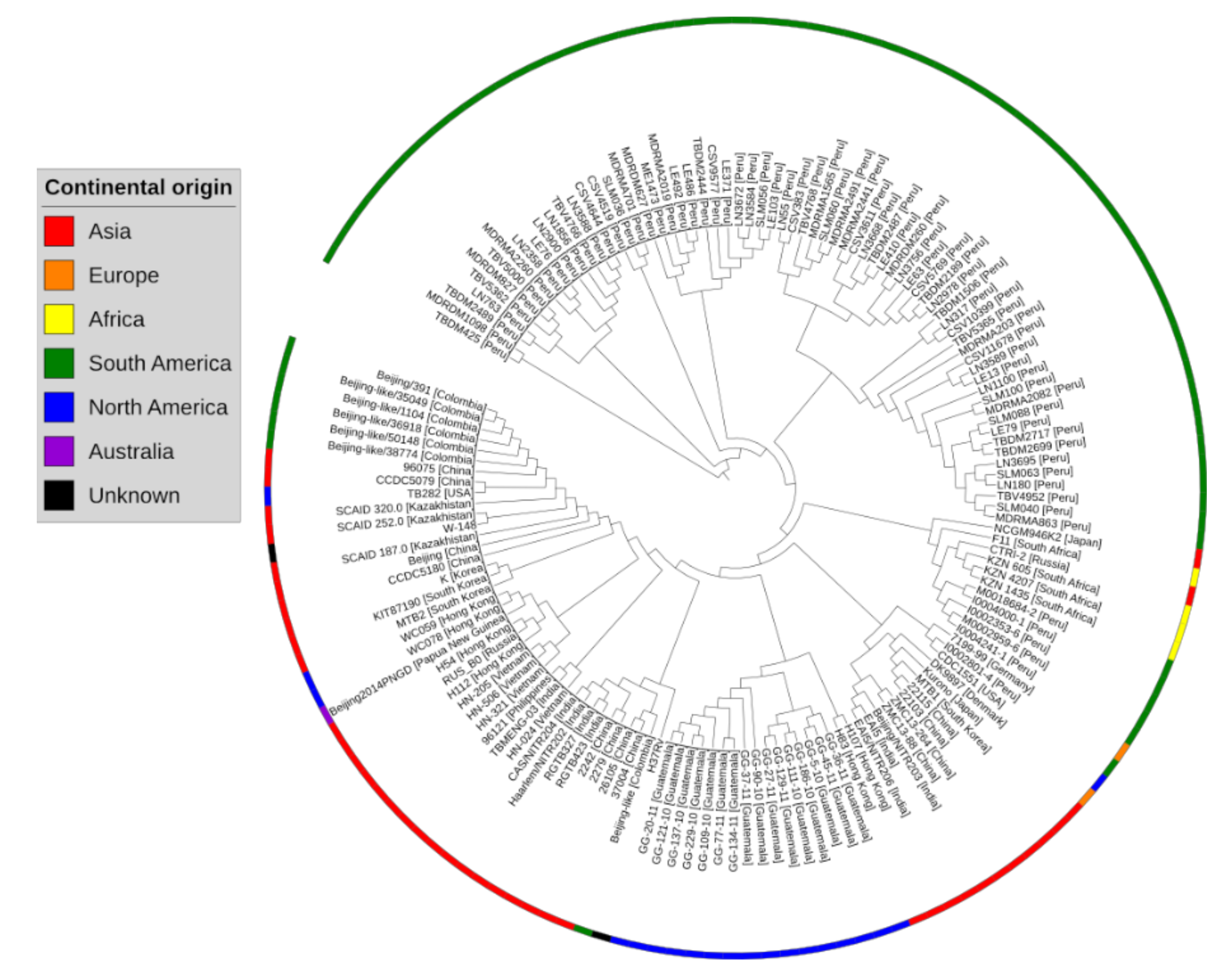
2.1. Pangenome and Pan-Phylogeny Analysis of Mycobacterium tuberculosis Genomes
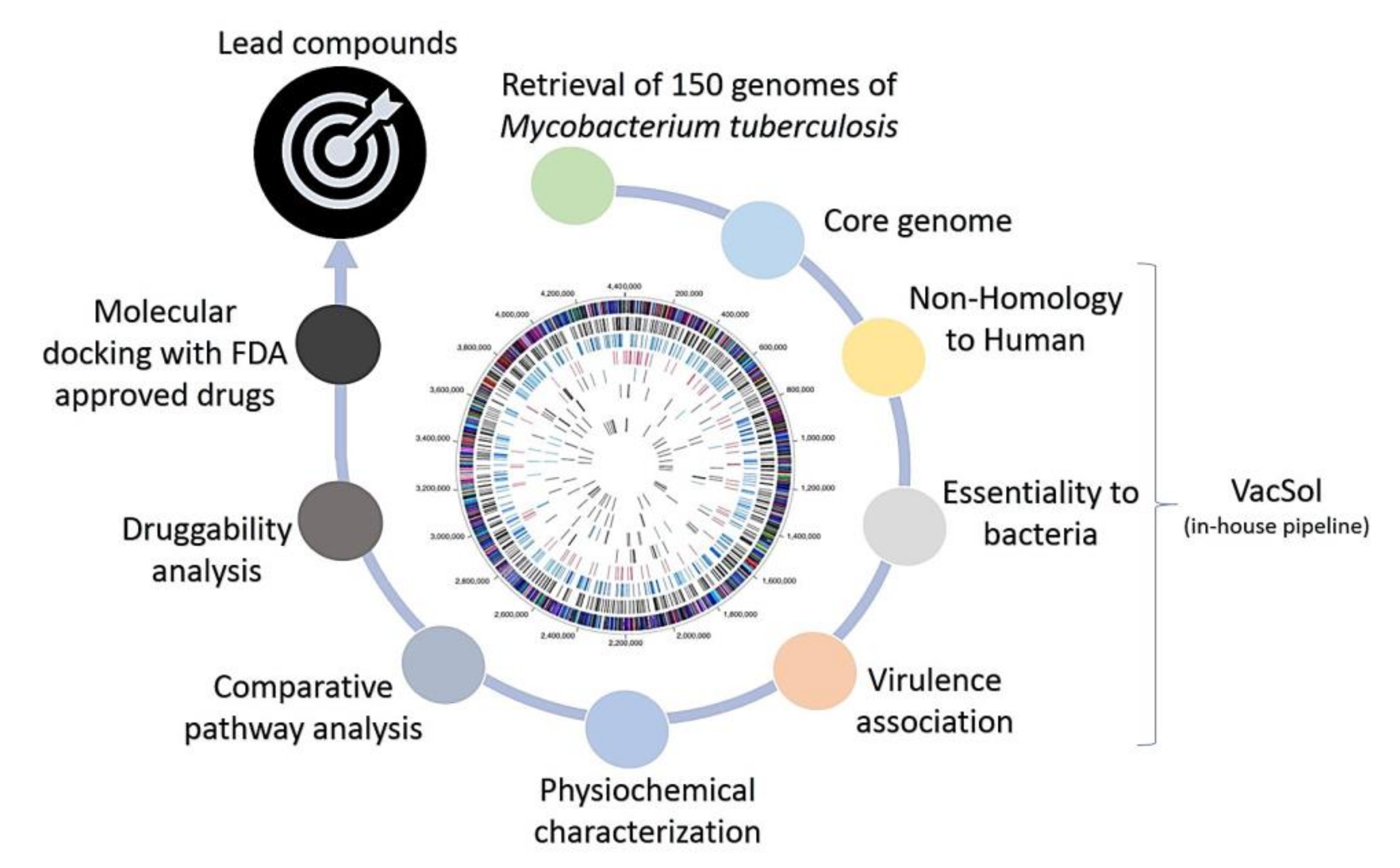
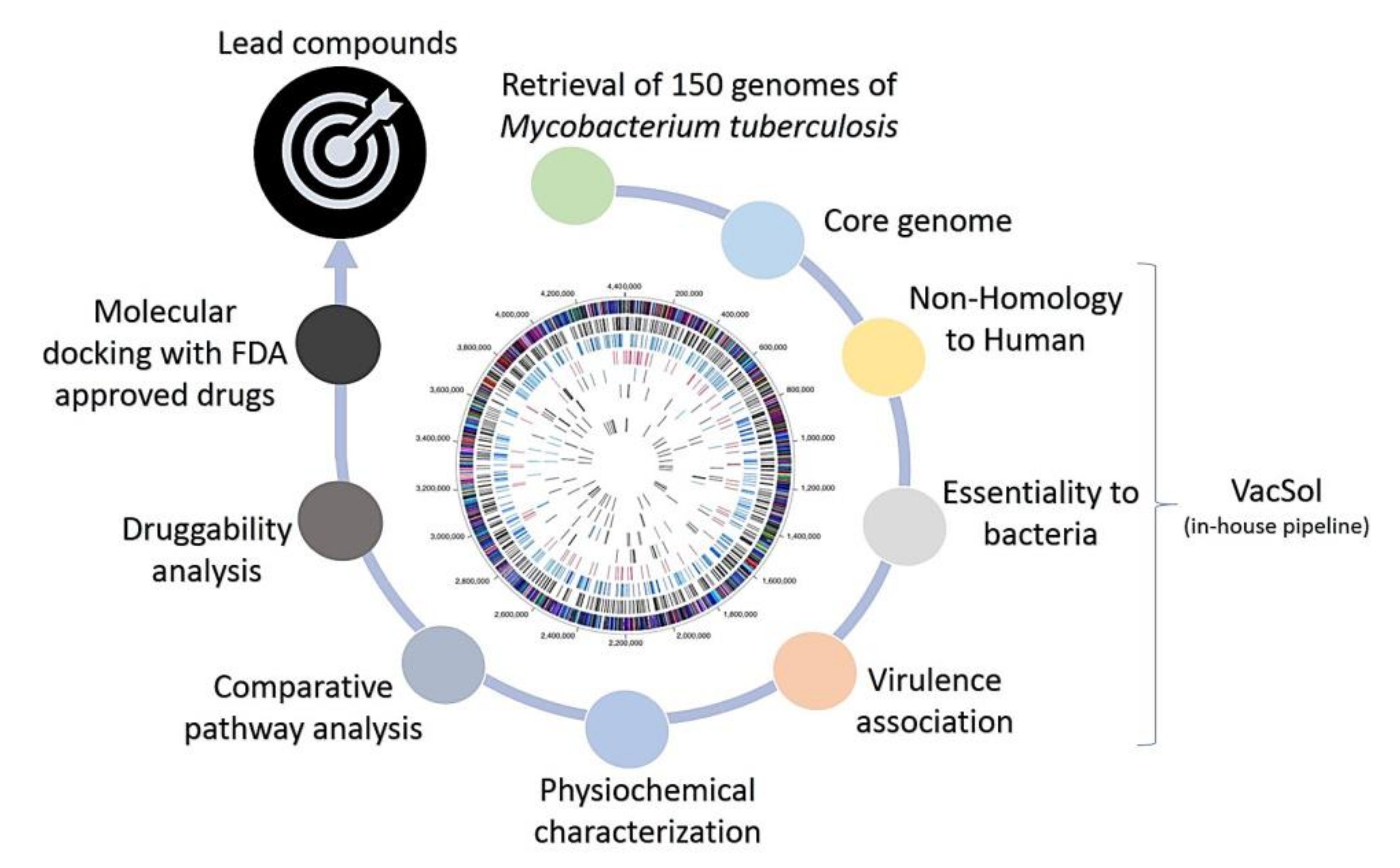
2.2. Subtractive Proteomics Revealed Putative Mycobacterium tuberculosis Drug Targets
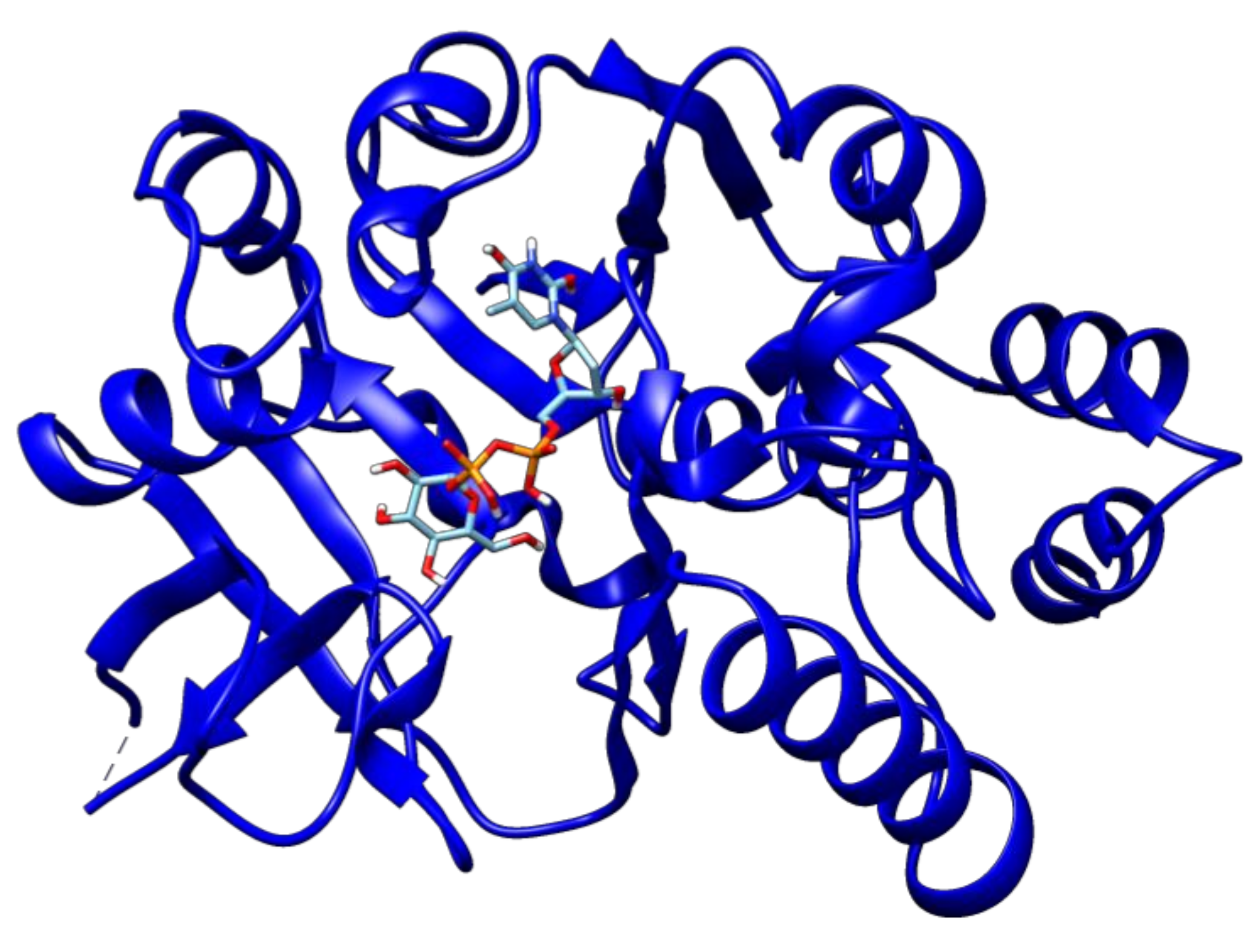
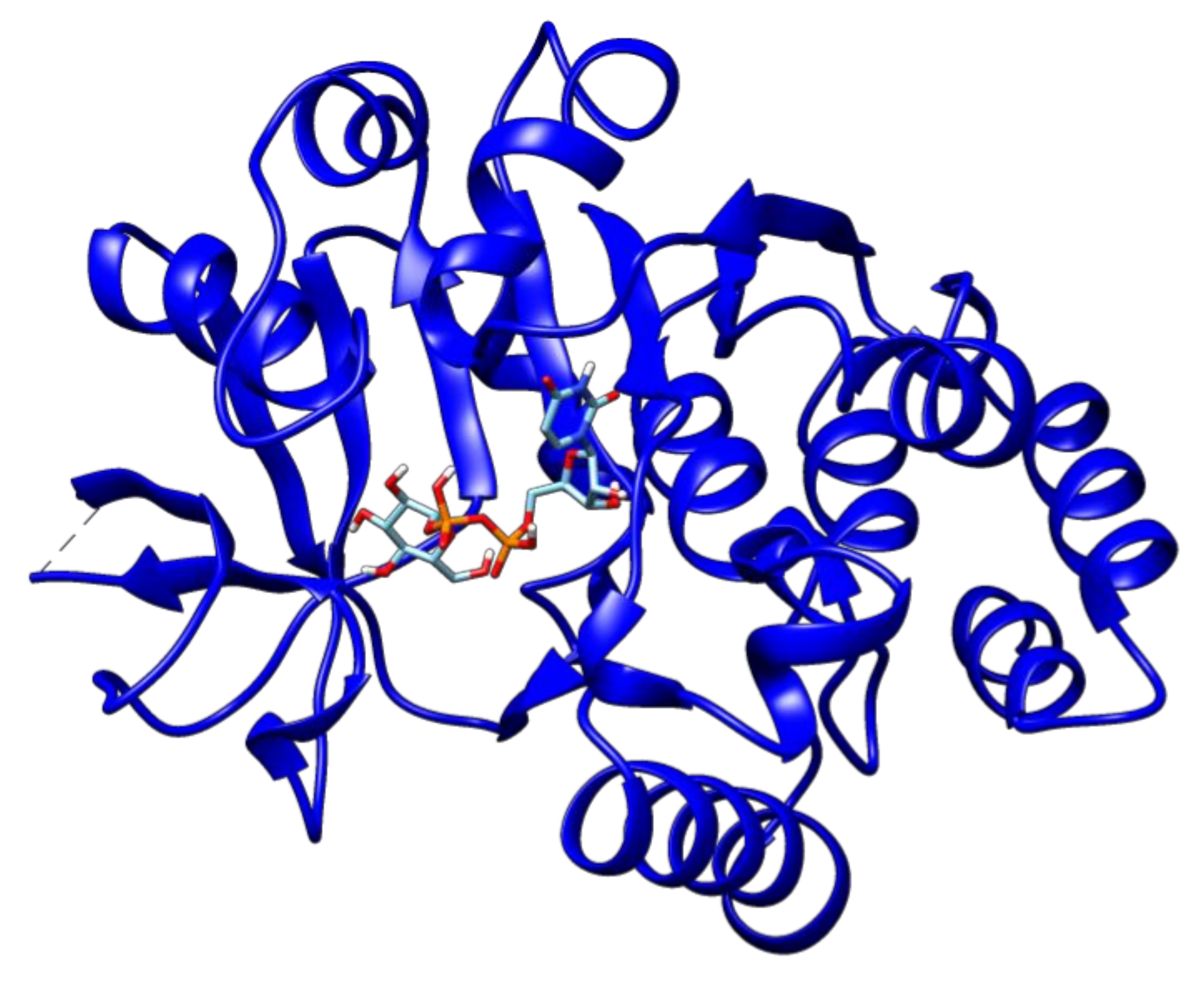
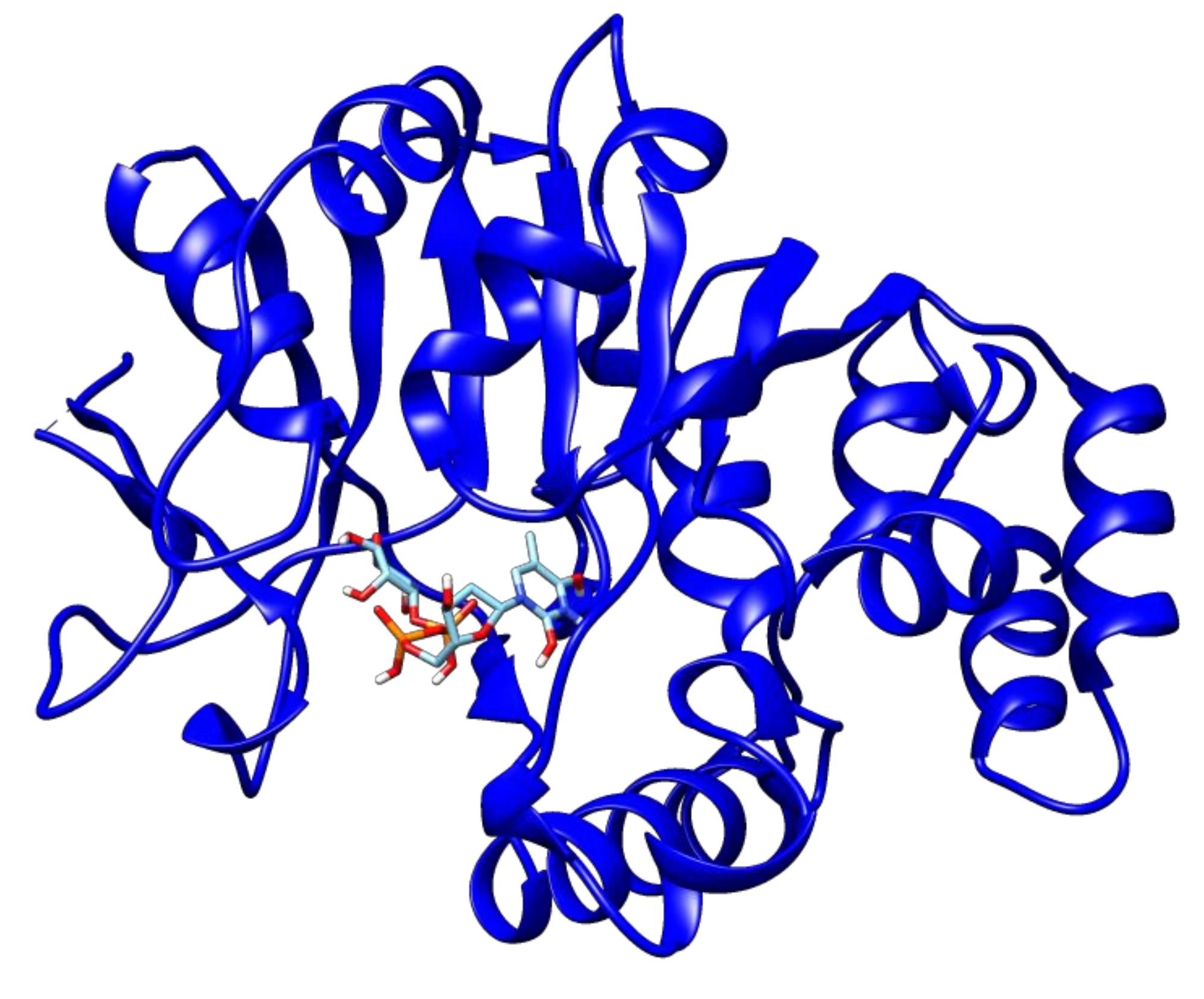
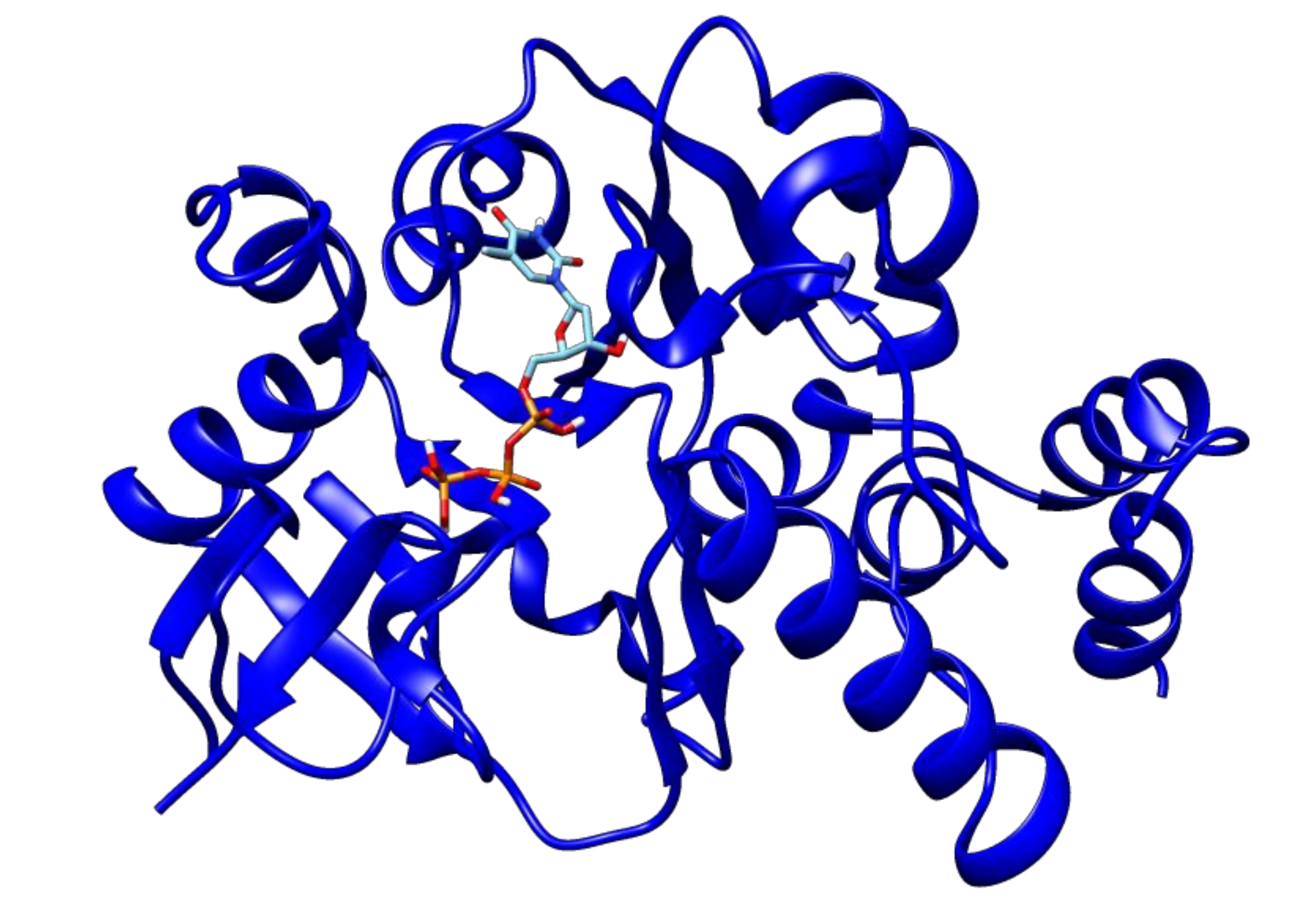
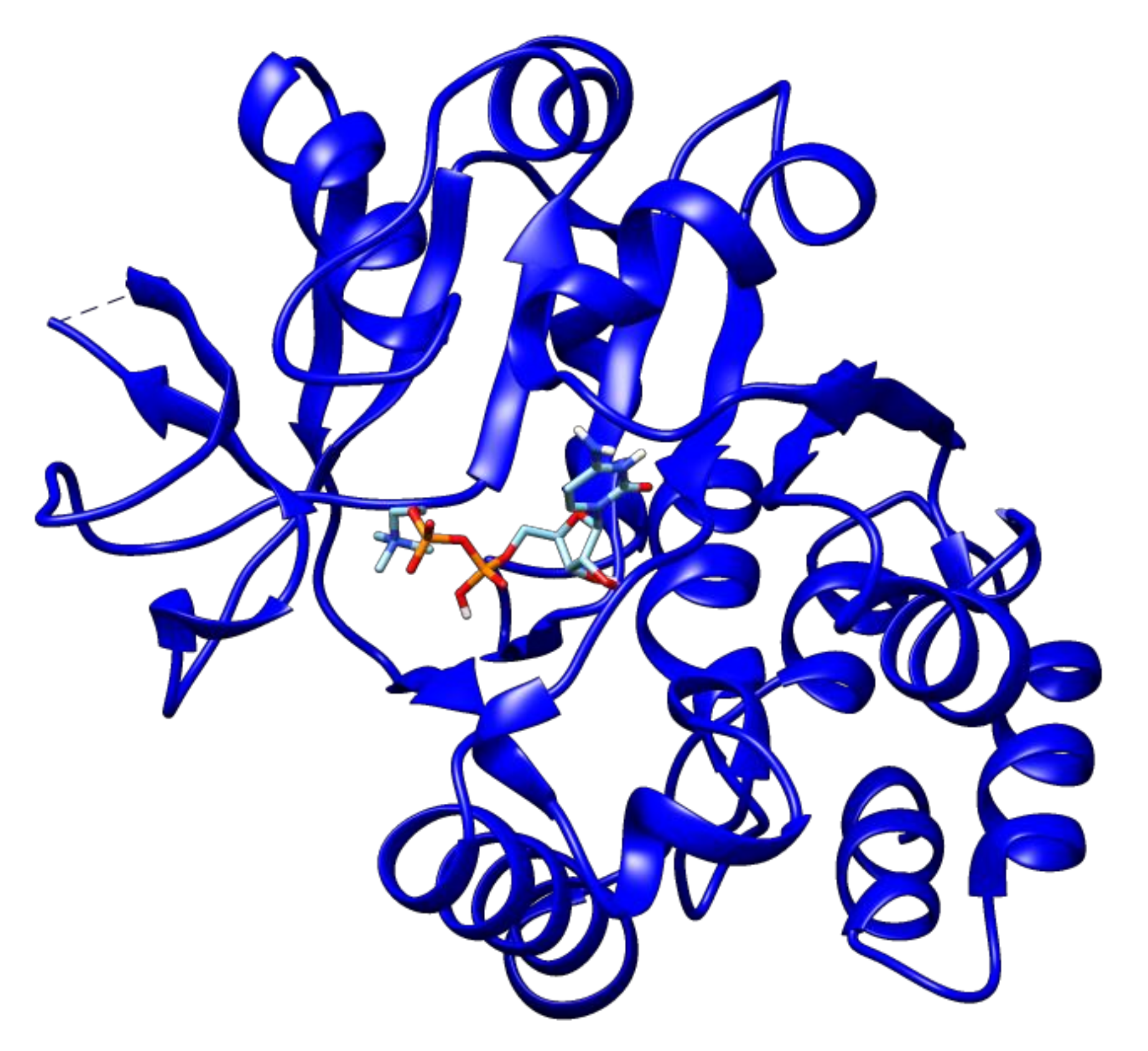
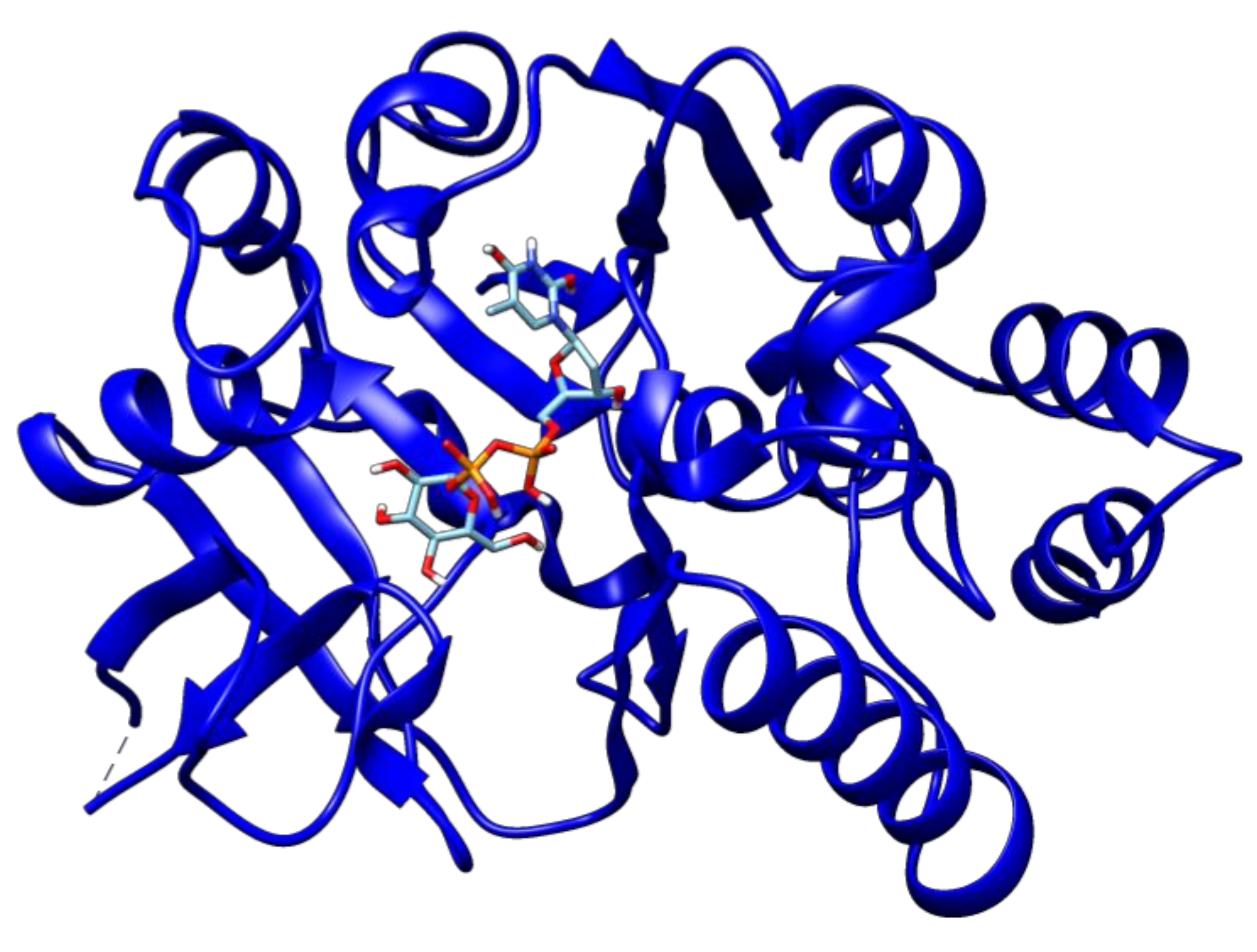
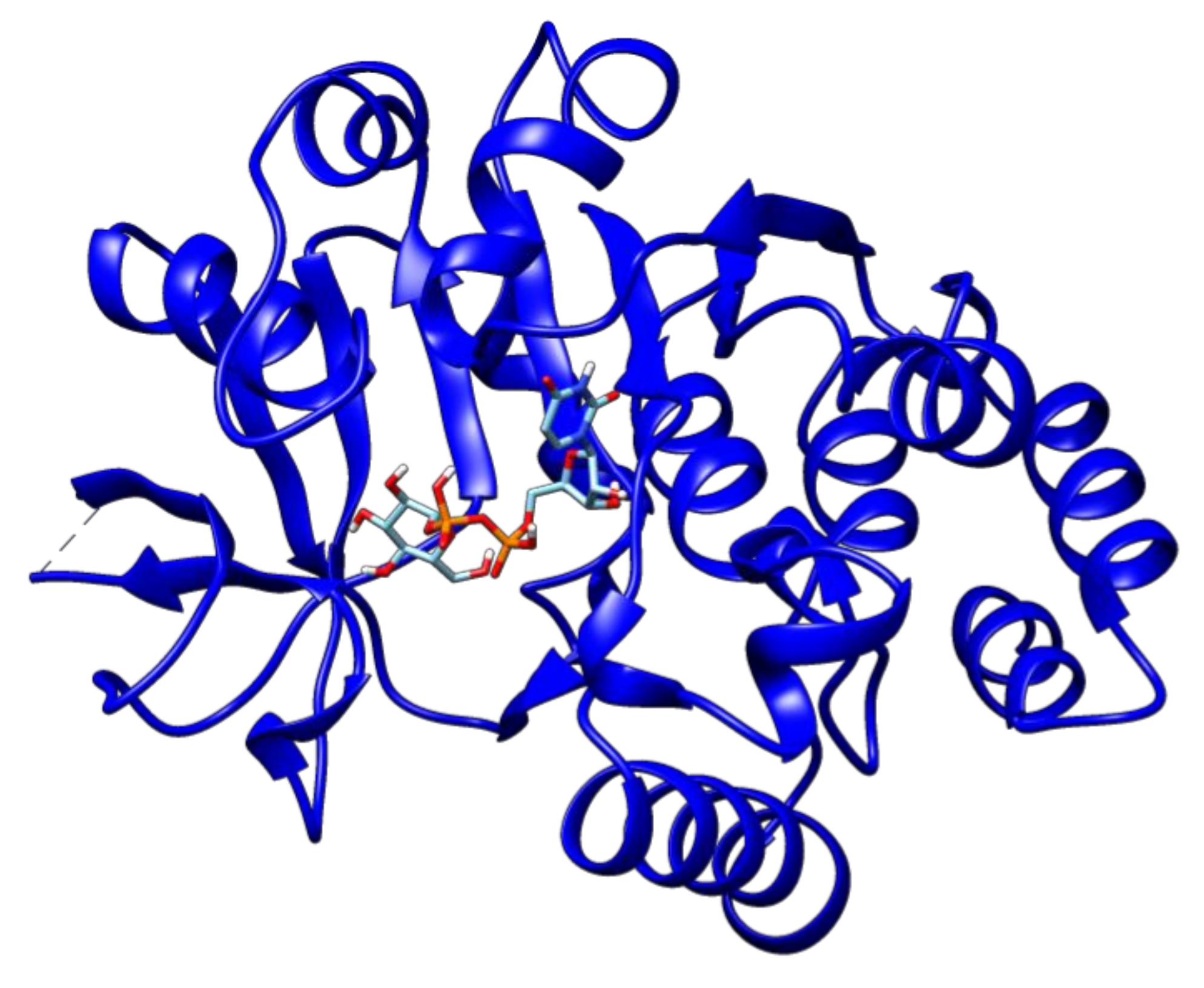
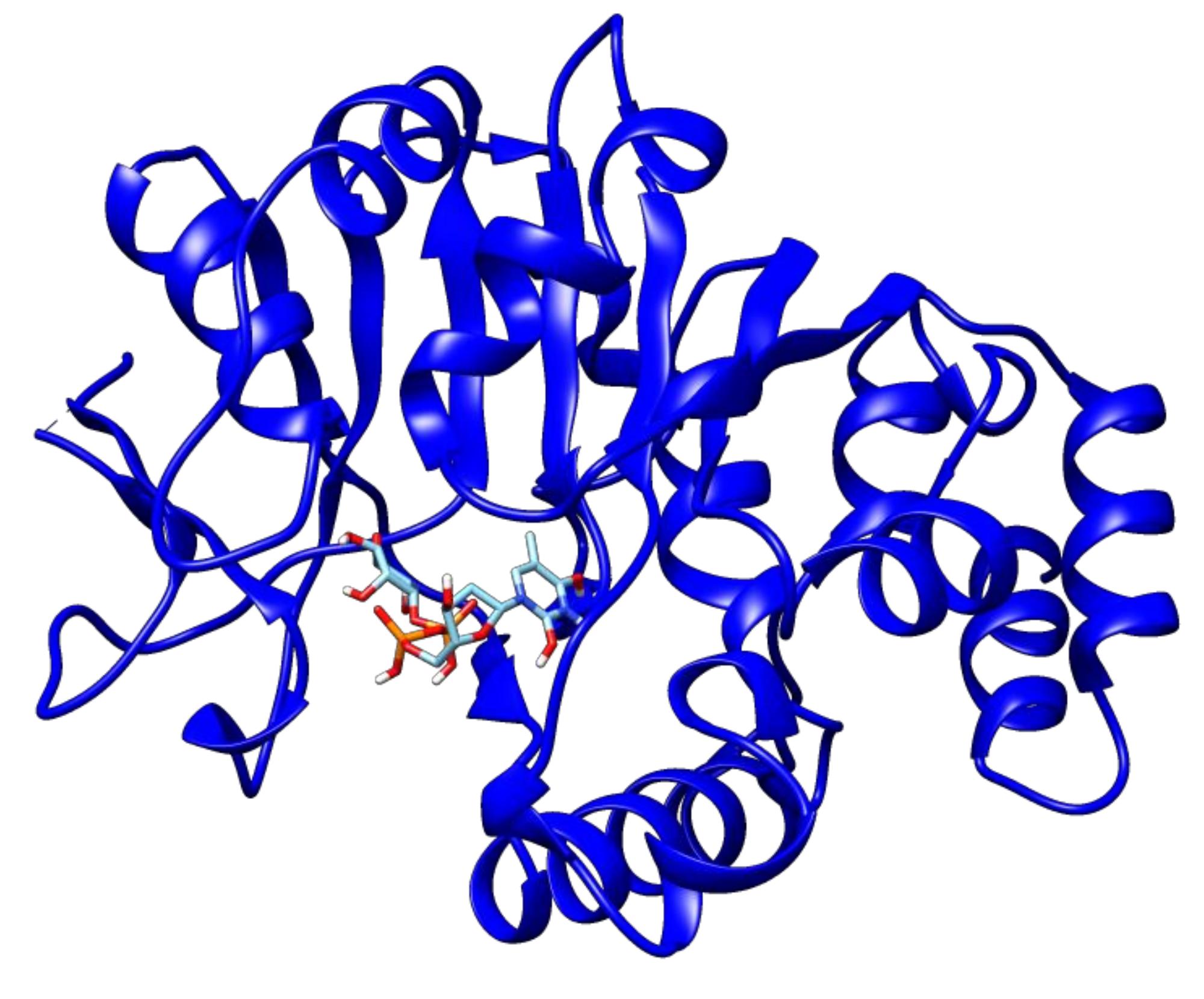
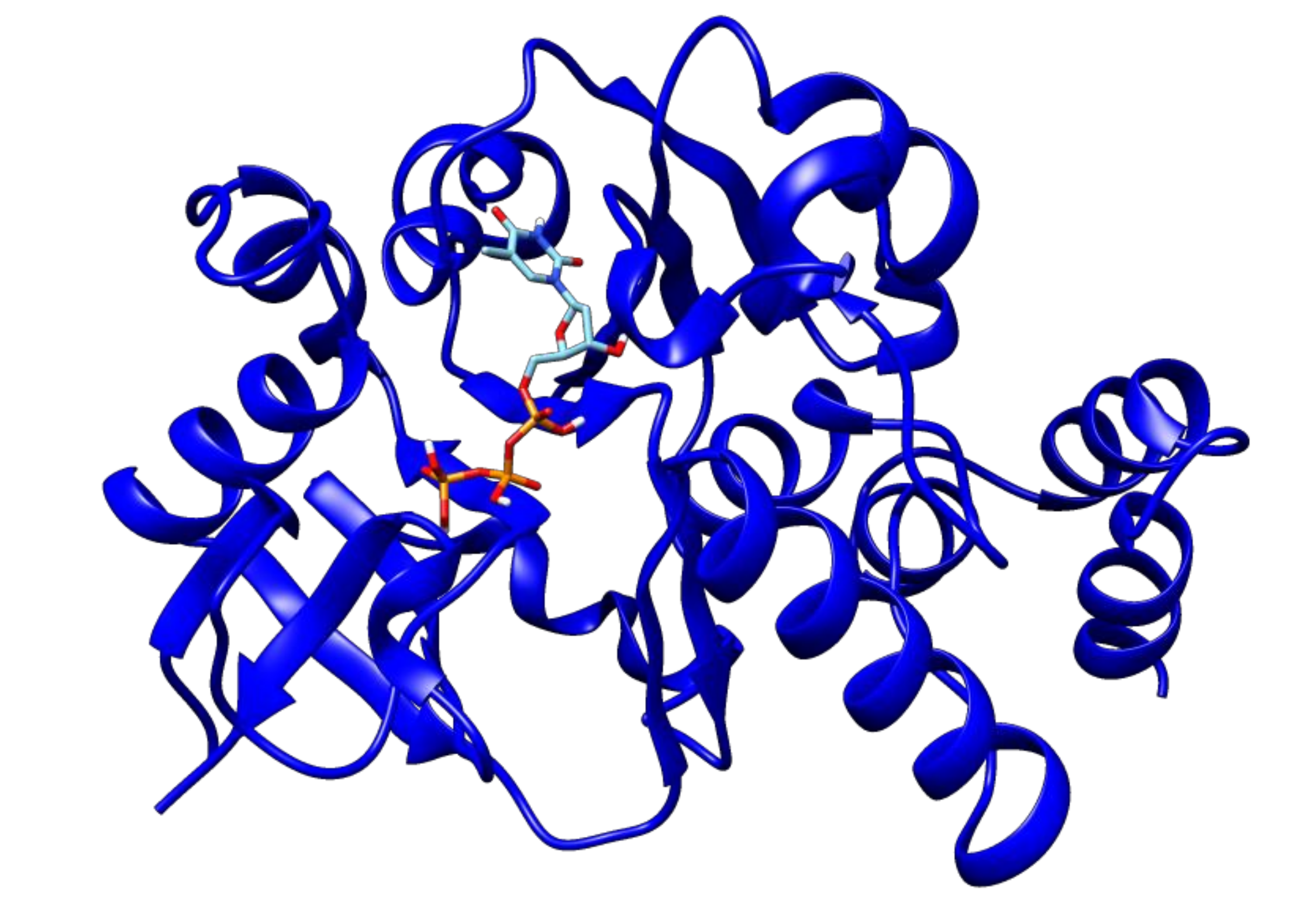
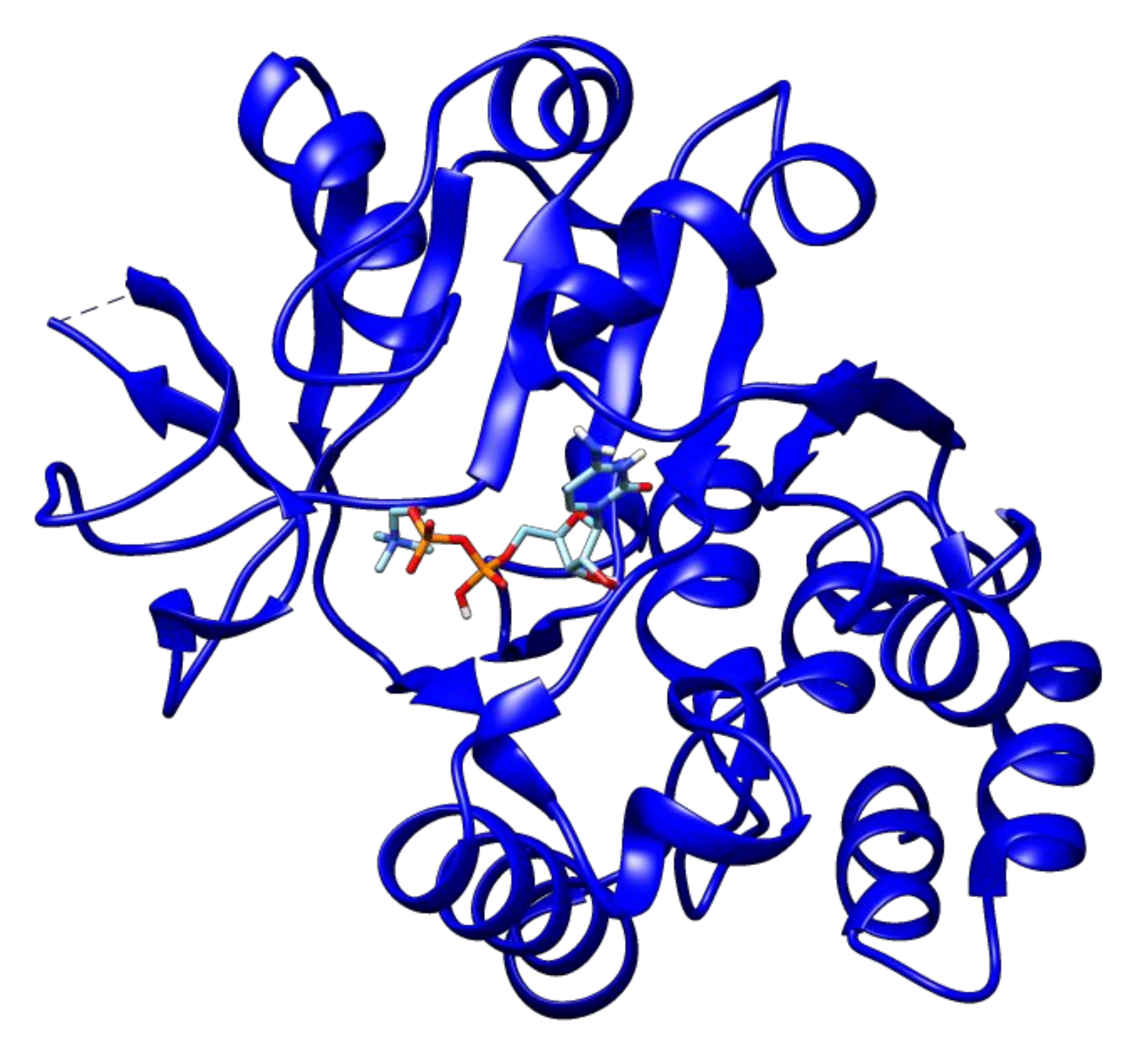
2.3. Docking Analyses of Drug Targets Revealed Potential Lead Compounds for Drug Discovery
3. Materials and Methods
3.1. Collection of Genomic Data
3.2. Pangenome Analysis of Mycobacterium tuberculosis Strains
3.3. Identification of Non-Host Homologous, Essential, and Virulence-Associated Proteins
3.4. Identification of Putative Mycobacterium tuberculosis Drug Targets
3.5. Molecular Docking of Putative Drug Targets with Drugs
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Gengenbacher, M.; Kaufmann, S.H.E. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, N.R.; Nunn, P.; Dheda, K.; Schaaf, H.S.; Zignol, M.; Van Soolingen, D. Multidrug-resistant and extensively drug-resistant tuberculosis: A threat to global control of tuberculosis. Lancet 2010, 375, 1830–1843. [Google Scholar] [CrossRef]
- Brindha, S.; Vincent, S.; Velmurugan, D.; Ananthakrishnan, D.; Sundaramurthi, J.C.; Gnanadoss, J.J. Bioinformatics approach to prioritize known drugs towards repurposing for tuberculosis. Med. Hypotheses 2017, 103, 39–45. [Google Scholar] [CrossRef]
- Cole, S.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537. [Google Scholar] [CrossRef]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Sundaramurthi, J.C.; Brindha, S.; Reddy, T.B.K.; Hanna, L.E. Informatics resources for tuberculosis—Towards drug discovery. Tuberculosis 2012, 92, 133–138. [Google Scholar] [CrossRef]
- Ekins, S.; Freundlich, J.S.; Choi, I.; Sarker, M.; Talcott, C. Computational databases, pathway and cheminformatics tools for tuberculosis drug discovery. Trends Microbiol. 2011, 19, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Karunakar, P.; Girija, C.R.; Krishnamurthy, V.; Krishna, V.; Shivakumar, K.V. In Silico antitubercular activity analysis of benzofuran and naphthofuran derivatives. Tuberc. Res. Treat. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Perryman, A.L.; Yu, W.; Wang, X.; Ekins, S.; Forli, S.; Li, S.-G. A virtual screen discovers novel, fragment-sized inhibitors of Mycobacterium tuberculosis InhA. J. Chem. Inf. Model 2015, 55, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Timo, G.O.; Reis, R.; de Melo, A.F.; Costa, T.V.L.; de Magalhães, P.O.; Homem-de-Mello, M. Predictive power of in Silico approach to evaluate chemicals against M. tuberculosis: A systematic review. Pharmaceuticals 2019, 12, 135. [Google Scholar] [CrossRef] [Green Version]
- Langer, T.; Wolber, G. Pharmacophore definition and 3D searches. Drug Discov. Today Technol. 2004, 1, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comas, I.; Chakravartti, J.; Small, P.M.; Galagan, J.; Niemann, S.; Kremer, K. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 2010, 42, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavvas, E.S.; Catoiu, E.; Mih, N.; Yurkovich, J.T.; Seif, Y.; Dillon, N. Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat. Commun. 2018, 9, 4306. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, A.; Morales, J.; Zambrano, M.M. Analysis of the genetic variation in Mycobacterium tuberculosis strains by multiple genome alignments. BMC Res. Notes 2008, 1, 110. [Google Scholar] [CrossRef] [Green Version]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef]
- Pang, X.; Cao, G.; Neuenschwander, P.F.; Haydel, S.E.; Hou, G.; Howard, S.T. The β-propeller gene Rv1057 of Mycobacterium tuberculosis has a complex promoter directly regulated by both the MprAB and TrcRS two-component systems. Tuberculosis 2011. [Google Scholar] [CrossRef] [Green Version]
- Haydel, S.E.; Benjamin, W.H.; Dunlap, N.E.; Clark-Curtiss, J.E. Expression, autoregulation, and DNA binding properties of the Mycobacterium tuberculosis TrcR response regulator. J. Bacteriol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.K.; Sarkar, I.; Sen, A. Herbal medicine meets bioinformatics for remedy of tuberculosis by Mycobacterium tuberculosis RGTB423. Int. J. Data Min. Bioinform. 2019. [Google Scholar] [CrossRef]
- Raman, K.; Yeturu, K.; Chandra, N. targetTB: A target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst. Biol. 2008, 2, 109. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Stern, R.J.; Scherman, M.S.; Vissa, V.D.; Yan, W.; Jones, V.C. Drug targeting Mycobacterium tuberculosis cell wall synthesis: Genetics of dTDP-rhamnose synthetic enzymes and development of a microtiter plate-based screen for inhibitors of conversion of dTDP-glucose to dTDP-rhamnose. Antimicrob. Agents Chemother. 2001, 45, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Barry, C.E., III. New horizons in the treatment of tuberculosis. Biochem. Pharmacol. 1997, 54, 1165–1172. [Google Scholar] [CrossRef]
- Gorla, P.; Plocinska, R.; Sarva, K.; Satsangi, A.T.; Pandeeti, E.; Donnelly, R. MtrA response regulator controls cell division and cell wall metabolism and affects susceptibility of mycobacteria to the first line antituberculosis drugs. Front. Microbiol. 2018. [Google Scholar] [CrossRef]
- Li, X.; Lv, X.; Lin, Y.; Zhen, J.; Ruan, C.; Duan, W. Role of two-component regulatory systems in intracellular survival of Mycobacterium tuberculosis. J. Cell Biochem. 2019, 120, 12197–12207. [Google Scholar] [CrossRef]
- Rifat, D.; Belchis, D.A.; Karakousis, P.C. SenX3-independent contribution of regX3 to Mycobacterium tuberculosis virulence. BMC Microbiol. 2014. [Google Scholar] [CrossRef] [Green Version]
- White, D.W.; Elliott, S.R.; Odean, E.; Bemis, L.T.; Tischler, A.D. Mycobacterium tuberculosis Pst/SenX3-RegX3 regulates membrane vesicle production independently of ESX-5 activity. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Freeman, Z.N.; Dorus, S.; Waterfield, N.R. The KdpD/KdpE two-component system: Integrating K+ homeostasis and virulence. PLoS Pathog. 2013, 9, e1003201. [Google Scholar] [CrossRef] [Green Version]
- Njoroge, J.W.; Gruber, C.; Sperandio, V. The interacting Cra and KdpE regulators are involved in the expression of multiple virulence factors in enterohemorrhagic Escherichia coli. J. Bacteriol. 2013, 195, 2499–2508. [Google Scholar] [CrossRef] [Green Version]
- Parker, C.T.; Russell, R.; Njoroge, J.W.; Jimenez, A.G.; Taussig, R.; Sperandio, V. Genetic and mechanistic analyses of the periplasmic domain of the enterohemorrhagic Escherichia coli QseC histidine sensor kinase. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [Green Version]
- Parish, T.; Smith, D.A.; Kendall, S.; Casali, N.; Bancroft, G.J.; Stoker, N.G. Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect. Immun. 2003, 71, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Alegado, R.A.; Chin, C.-Y.; Monack, D.M.; Tan, M.-W. The two-component sensor kinase KdpD is required for Salmonella typhimurium colonization of Caenorhabditis elegans and survival in macrophages. Cell Microbiol. 2011, 13, 1618–1637. [Google Scholar] [CrossRef]
- Xue, T.; You, Y.; Hong, D.; Sun, H.; Sun, B. The Staphylococcus aureus KdpDE two-component system couples extracellular K+ sensing and Agr signaling to infection programming. Infect. Immun. 2011, 79, 2154–2167. [Google Scholar] [CrossRef] [Green Version]
- Feinbaum, R.L.; Urbach, J.M.; Liberati, N.T.; Djonovic, S.; Adonizio, A.; Carvunis, A.-R. Genome-wide identification of Pseudomonas aeruginosa virulence-related genes using a Caenorhabditis elegans infection model. PLoS Pathog. 2012, 8, e1002813. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, S.; Glaser, L. The enzymic synthesis of thymidine-linked sugars. I. Thymidine diphosphate glucose. J. Biol. Chem. 1961, 56, 184–185. [Google Scholar]
- Brown, H.A.; Thoden, J.B.; Tipton, P.A.; Holden, H.M. The structure of glucose-1-phosphate thymidylyltransferase from Mycobacterium tuberculosis reveals the location of an essential magnesium ion in the RmlA-type enzymes. Protein Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Xin, Y.; Dong, X.; Ma, Y. An rmlA gene encoding D-glucose-1-phosphate thymidylyltransferase is essential for mycobacterial growth. FEMS Microbiol. Lett. 2007. [Google Scholar] [CrossRef]
- Kantardjieff, K.A.; Kim, C.-Y.; Naranjo, C.; Waldo, G.S.; Lekin, T.; Segelke, B.W. Mycobacterium tuberculosis RmlC epimerase (Rv3465): A promising drug-target structure in the rhamnose pathway. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 895–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazir, Z.; Afridi, S.G.; Shah, M.; Shams, S.; Khan, A. Reverse vaccinology and subtractive genomics-based putative vaccine targets identification for Burkholderia pseudomallei Bp1651. Microb. Pathog. 2018, 125, 219–229. [Google Scholar]
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.-T.; Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014, 42, D574–D580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.E.; Smith, J.; Lam, M.; Zemla, A.; Dyer, M.D.; Slezak, T. MvirDB—A microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 2007, 35, D391–D394. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W. Bacterial pathogenesis. In Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 571–607. [Google Scholar]
- Chawley, P.; Samal, H.B.; Prava, J.; Suar, M.; Mahapatra, R.K. Comparative genomics study for identification of drug and vaccine targets in Vibrio cholerae: MurA ligase as a case study. Genomics 2014, 103, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Azam, S.S.; Shamim, A. An insight into the exploration of druggable genome of Streptococcus gordonii for the identification of novel therapeutic candidates. Genomics 2014, 104, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Mondal, S.I.; Ferdous, S.; Jewel, N.A.; Akter, A.; Mahmud, Z.; Islam, M.M. Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157: H7: An in silico approach. Adv. Appl. Bioinforma. Chem. AABC 2015, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Humana Press: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Rv Locus Number | KEGG Orthology | Metabolic Pathway(s) |
|---|---|---|---|
| dTDP-4-dehydrorhamnose reductase | Rv3266c | K00067 | ko00521 Streptomycin biosynthesis ko00523 Polyketide sugar unit biosynthesis ko00541 O-Antigen nucleotide sugar biosynthesis ko01100 Metabolic pathways ko01110 Biosynthesis of secondary metabolites |
| glucose-1-phosphate thymidylyltransferase | Rv0334 | K00973 | ko00521 Streptomycin biosynthesis ko00523 Polyketide sugar unit biosynthesis ko00525 Acarbose and validamycin biosynthesis ko00541 O-Antigen nucleotide sugar biosynthesis ko01100 Metabolic pathways ko01110 Biosynthesis of secondary metabolites |
| two-component system regulator trcR | Rv1033c | K07672 | ko02020 Two-component system |
| two-component system regulator mtrA | Rv3246c | K07670 | ko02020 Two-component system |
| two-component system regulator regX3 | Rv0491 | K07776 | ko02020 Two-component system |
| two-component system regulator kdpE | Rv1027c | K07667 | ko02020 Two-component system ko02024 Quorum sensing |
| two-component system regulator devR | Rv3133c | K07695 | ko02020 Two-component system |
| dTDP-4-dehydrorhamnose 3,5-epimerase | Rv3465 | K01790 | ko00521 Streptomycin biosynthesis ko00523 Polyketide sugar unit biosynthesis ko00541 O-Antigen nucleotide sugar biosynthesis ko01100 Metabolic pathways ko01110 Biosynthesis of secondary metabolites |
| Target | Rv Locus Number | Ligand | Binding Energy |
|---|---|---|---|
| Glucose-1-phosphate thymidylyltransferase | Rv0334 | 2′-Deoxy-Thymidine-Beta-l-Rhamnose | −9.1 |
| 2′deoxy-Thymidine-5′-Diphospho-Alpha-d-Glucose | −10.1 | ||
| Alpha-d-Glucose-1-Phosphate | −5.8 | ||
| Citicoline | −8.4 | ||
| Citric acid | −5.7 | ||
| Thymidine-5′-Triphosphate | −8.9 | ||
| Thymidine | −7.1 | ||
| Thymidine monophosphate | −7.7 | ||
| Uridine diphosphate glucose | −9.8 | ||
| DNA-binding response regulator | Rv1027c | Phosphoaspartate | −5.2 |
| Guanosine-5′-Monophosphate | −7.9 | ||
| AlphaBeta-Methyleneadenosine-5′-Triphosphate | −7.3 | ||
| Adenosine-5′-Rp-Alpha-Thio-Triphosphate | −6.8 | ||
| 2-Hydroxyestradiol | −7.9 | ||
| dTDP-4-dehydrorhamnose 3,5-epimerase | Rv3465 | 2′deoxy-Thymidine-5′-Diphospho-Alpha-d-Glucose | −7.2 |
| 3′-O-Acetylthymidine-5′-Diphosphate | −7.1 | ||
| d-tartaric acid | −4.6 | ||
| SS-(2-Hydroxyethyl)Thiocysteine | −4.6 | ||
| Thymidine_monophosphate | −6.8 | ||
| Thymidine-5′-diphospho-beta-d-xylose | −6.7 | ||
| DNA-binding response regulator TrcR | Rv1033c | S-Methyl Phosphocysteine | −4.6 |
| Phosphoaspartate | −4.8 | ||
| Guanosine-5′-Monophosphate | −7.2 | ||
| Glycerine | −4 | ||
| AlphaBeta-Methyleneadenosine-5′-Triphosphate | −7.4 | ||
| Adenosine-5′-Rp-Alpha-Thio-Triphosphate | −7.6 | ||
| 3-Aminosuccinimide | −4.5 | ||
| 2-Hydroxyestradiol | −7.1 | ||
| DNA-binding response regulator RegX3 | Rv0491 | 2-Hydroxyestradiol | −7.1 |
| 3-Aminosuccinimide | −4.4 | ||
| Adenosine-5′-Rp-Alpha-Thio-Triphosphate | −6.7 | ||
| AlphaBeta-Methyleneadenosine-5′-Triphosphate | −6.6 | ||
| Glycerine | −3.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dar, H.A.; Zaheer, T.; Ullah, N.; Bakhtiar, S.M.; Zhang, T.; Yasir, M.; Azhar, E.I.; Ali, A. Pangenome Analysis of Mycobacterium tuberculosis Reveals Core-Drug Targets and Screening of Promising Lead Compounds for Drug Discovery. Antibiotics 2020, 9, 819. https://doi.org/10.3390/antibiotics9110819
Dar HA, Zaheer T, Ullah N, Bakhtiar SM, Zhang T, Yasir M, Azhar EI, Ali A. Pangenome Analysis of Mycobacterium tuberculosis Reveals Core-Drug Targets and Screening of Promising Lead Compounds for Drug Discovery. Antibiotics. 2020; 9(11):819. https://doi.org/10.3390/antibiotics9110819
Chicago/Turabian StyleDar, Hamza Arshad, Tahreem Zaheer, Nimat Ullah, Syeda Marriam Bakhtiar, Tianyu Zhang, Muhammad Yasir, Esam I. Azhar, and Amjad Ali. 2020. "Pangenome Analysis of Mycobacterium tuberculosis Reveals Core-Drug Targets and Screening of Promising Lead Compounds for Drug Discovery" Antibiotics 9, no. 11: 819. https://doi.org/10.3390/antibiotics9110819
APA StyleDar, H. A., Zaheer, T., Ullah, N., Bakhtiar, S. M., Zhang, T., Yasir, M., Azhar, E. I., & Ali, A. (2020). Pangenome Analysis of Mycobacterium tuberculosis Reveals Core-Drug Targets and Screening of Promising Lead Compounds for Drug Discovery. Antibiotics, 9(11), 819. https://doi.org/10.3390/antibiotics9110819