Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates

 , ,
, ,
Abstract
1. Introduction
2. Results
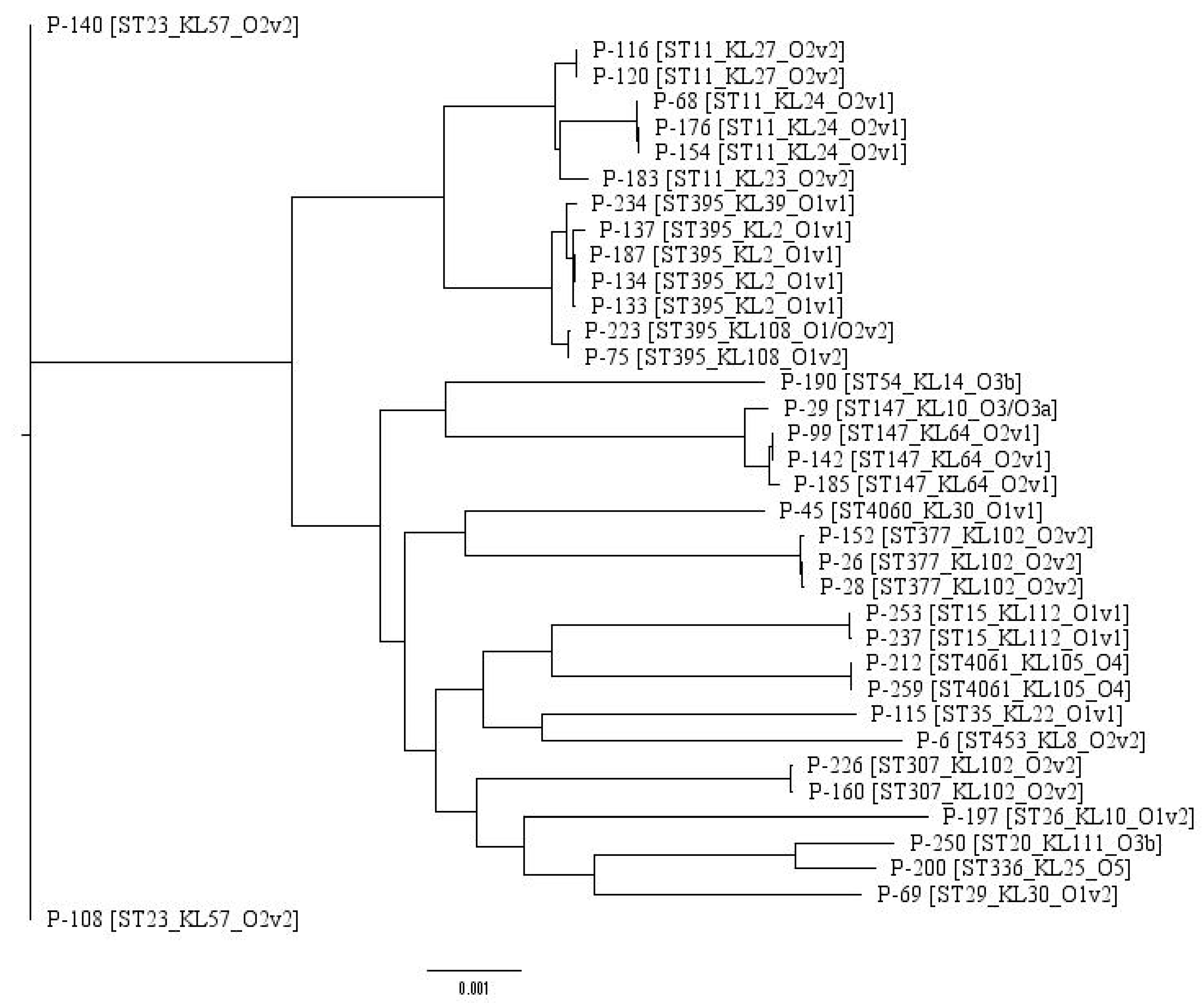
2.1. Typing and Classification
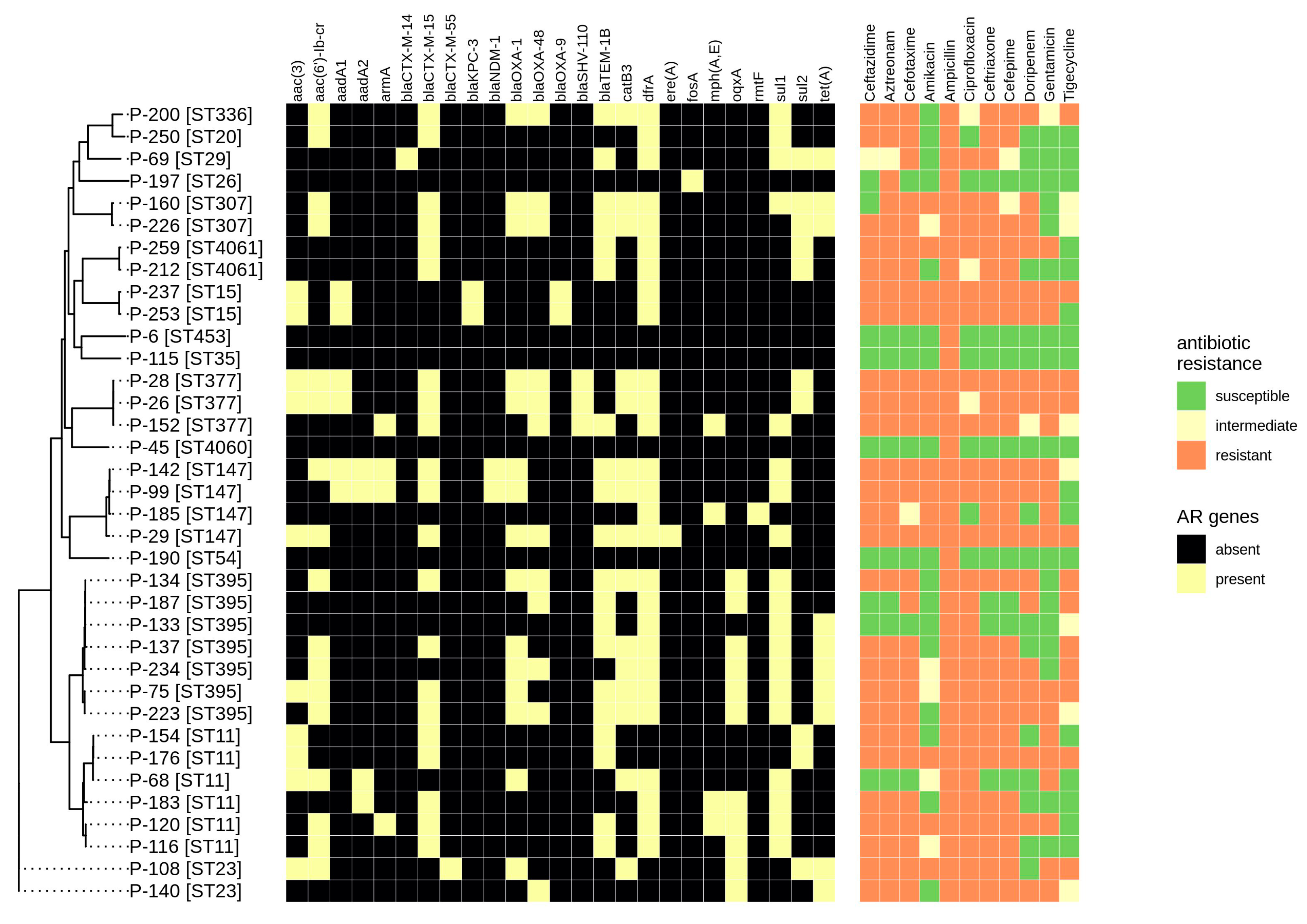
2.2. Antibiotic Resistance Determination
2.3. Plasmids
2.4. Virulence Genes
3. Discussion
4. Materials and Methods
4.1. DNA Isolation, Sequencing and Genome Assembly
4.2. Ethical Statement
4.3. Determination of Antibiotic Susceptibility
4.4. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Marahiel, M.A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef] [PubMed]
- Turton, J.F.; Payne, Z.; Coward, A.; Hopkins, K.L.; Turton, J.A.; Doumith, M.; Woodford, N. Virulence genes in isolates of Klebsiella pneumoniae from the UK during 2016, including among carbapenemase gene-positive hypervirulent K1-ST23 and ‘non-hypervirulent’ types ST147, ST15 and ST383. J. Med. Microbiol. 2018, 67, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, B.S.; Chun, J.; Yong, J.H.; Lee, Y.S.; Yoo, J.S.; Yong, D.; Hong, S.G.; D’Souza, R.; Thomson, K.S.; et al. Clonality and Resistome analysis of KPC-producing Klebsiella pneumoniae strain isolated in Korea using whole genome sequencing. Biomed. Res. Int. 2014, 2014, 352862. [Google Scholar] [CrossRef]
- Karlsson, M.; Stanton, R.A.; Ansari, U.; McAllister, G.; Chan, M.Y.; Sula, E.; Grass, J.E.; Duffy, N.; Anacker, M.L.; Witwer, M.L.; et al. Identification of a Carbapenemase-Producing Hypervirulent Klebsiella pneumoniae Isolate in the United States. Antimicrob. Agents Chemother. 2019, 63, e00519-19. [Google Scholar] [CrossRef]
- Pitout, J.D.; Nordmann, P.; Poirel, L. Carbapenemase-Producing Klebsiella pneumoniae, a Key Pathogen Set for Global Nosocomial Dominance. Antimicrob. Agents Chemother. 2015, 59, 5873–5884. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Lin, T.L.; Pan, Y.J.; Wang, Y.P.; Lin, Y.T.; Wang, J.T. Colistin resistance mechanisms in Klebsiella pneumoniae strains from Taiwan. Antimicrob. Agents Chemother. 2015, 59, 2909–2913. [Google Scholar] [CrossRef]
- Ah, Y.M.; Kim, A.J.; Lee, J.Y. Colistin resistance in Klebsiella pneumoniae. Int. J. Antimicrob. Agents 2014, 44, 8–15. [Google Scholar] [CrossRef]
- Tamma, P.D.; Fan, Y.; Bergman, Y.; Pertea, G.; Kazmi, A.Q.; Lewis, S.; Carroll, K.C.; Schatz, M.C.; Timp, W.; Simner, P.J. Applying Rapid Whole-Genome Sequencing to Predict Phenotypic Antimicrobial Susceptibility Testing Results among Carbapenem-Resistant Klebsiella pneumoniae Clinical Isolates. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Martin, J.; Phan, H.T.T.; Findlay, J.; Stoesser, N.; Pankhurst, L.; Navickaite, I.; De Maio, N.; Eyre, D.W.; Toogood, G.; Orsi, N.M.; et al. Covert dissemination of carbapenemase-producing Klebsiella pneumoniae (KPC) in a successfully controlled outbreak: Long- and short-read whole-genome sequencing demonstrate multiple genetic modes of transmission. J. Antimicrob. Chemother. 2017, 72, 3025–3034. [Google Scholar] [CrossRef]
- Agyepong, N.; Govinden, U.; Owusu-Ofori, A.; Amoako, D.G.; Allam, M.; Janice, J.; Pedersen, T.; Sundsfjord, A.; Essack, S. Genomic characterization of multidrug-resistant ESBL-producing Klebsiella pneumoniae isolated from a Ghanaian teaching hospital. Int. J. Infect. Dis. 2019, 85, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, S.; Crawford, M.A.; Lascols, C.; Timme, R.E.; Anderson, K.; Hodge, D.R.; Fisher, D.J.; Pillai, S.P.; Morse, S.A.; Khan, E.; et al. Resistome of carbapenem- and colistin-resistant Klebsiella pneumoniae clinical isolates. PLoS ONE 2018, 13, e0198526. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Diancourt, L.; Passet, V.; Verhoef, J.; Grimont, P.A.; Brisse, S. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J. Clin. Microbiol. 2005, 43, 4178–4182. [Google Scholar] [CrossRef] [PubMed]
- Klebsiella Sequence Typing. Available online: http://bigsdb.pasteur.fr (accessed on 10 April 2020).
- Wick, R.R.; Heinz, E.; Holt, K.E.; Wyres, K.L. Kaptive Web: User-Friendly Capsule and Lipopolysaccharide Serotype Prediction for Klebsiella Genomes. J. Clin. Microbiol. 2018, 56, e00197-18. [Google Scholar] [CrossRef]
- Shelenkov, A.; Korotkov, E. Search of regular sequences in promoters from eukaryotic genomes. Comput. Biol. Chem. 2009, 33, 196–204. [Google Scholar] [CrossRef]
- Compain, F.; Babosan, A.; Brisse, S.; Genel, N.; Audo, J.; Ailloud, F.; Kassis-Chikhani, N.; Arlet, G.; Decre, D. Multiplex PCR for detection of seven virulence factors and K1/K2 capsular serotypes of Klebsiella pneumoniae. J. Clin. Microbiol. 2014, 52, 4377–4380. [Google Scholar] [CrossRef]
- Hennequin, C.; Robin, F. Correlation between antimicrobial resistance and virulence in Klebsiella pneumoniae. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 333–341. [Google Scholar] [CrossRef]
- Frasson, I.; Cavallaro, A.; Bergo, C.; Richter, S.N.; Palu, G. Prevalence of aac(6′)-Ib-cr plasmid-mediated and chromosome-encoded fluoroquinolone resistance in Enterobacteriaceae in Italy. Gut Pathog. 2011, 3, 12. [Google Scholar] [CrossRef]
- Litrup, E.; Kiil, K.; Hammerum, A.M.; Roer, L.; Nielsen, E.M.; Torpdahl, M. Plasmid-borne colistin resistance gene mcr-3 in Salmonella isolates from human infections, Denmark 2009–2017. Eurosurveillance 2017, 22, 30587. [Google Scholar] [CrossRef]
- Greig, D.R.; Dallman, T.J.; Hopkins, K.L.; Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microb. Genom. 2018, 4, e000213. [Google Scholar] [CrossRef] [PubMed]
- Ragupathi, N.K.D.; Bakthavatchalam, Y.D.; Mathur, P.; Pragasam, A.K.; Walia, K.; Ohri, V.C.; Veeraraghavan, B. Plasmid profiles among some ESKAPE pathogens in a tertiary care centre in south India. Indian J. Med. Res. 2019, 149, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Falgenhauer, L.; Waezsada, S.-E.; Yao, Y.; Imirzalioglu, C.; Käsbohrer, A.; Roesler, U.; Michael, G.B.; Schwarz, S.; Werner, G.; Kreienbrock, L.; et al. Colistin resistance gene mcr-1 in extended-spectrum β-lactamase-producing and carbapenemase-producing Gram-negative bacteria in Germany. Lancet Infect. Dis. 2016, 16, 282–283. [Google Scholar] [CrossRef]
- Li, B.; Zhao, Y.; Liu, C.; Chen, Z.; Zhou, D. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol. 2014, 9, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Rouhi, S.; Ahmadi, A.; Ramazanzadeh, R.; Kalani, M.T.; Shakib, P. Molecular detection of virulence genes in Klebsiella Pneumoniae clinical isolates from Kurdistan Province, Iran. Biomed. Res. Ther. 2018, 5, 2581–2589. [Google Scholar] [CrossRef]
- Fursova, N.K.; Astashkin, E.I.; Gabrielyan, N.I.; Novikova, T.S.; Fedyukina, G.N.; Kubanova, M.K.; Esenova, N.M.; Sharapchenko, S.O.; Volozhantsev, N.V. Emergence of Five Genetic Lines ST395(NDM-1), ST13(OXA-48), ST3346(OXA-48), ST39(CTX-M-14), and Novel ST3551(OXA-48) of Multidrug-Resistant Clinical Klebsiella pneumoniae in Russia. Microb. Drug Resist. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fursova, N.K.; Astashkin, E.I.; Knyazeva, A.I.; Kartsev, N.N.; Leonova, E.S.; Ershova, O.N.; Alexandrova, I.A.; Kurdyumova, N.V.; Sazikina, S.Y.; Volozhantsev, N.V.; et al. The spread of bla OXA-48 and bla OXA-244 carbapenemase genes among Klebsiella pneumoniae, Proteus mirabilis and Enterobacter spp. isolated in Moscow, Russia. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Ageevets, V.A.; Partina, I.V.; Lisitsyna, E.S.; Ilina, E.N.; Lobzin, Y.V.; Shlyapnikov, S.A.; Sidorenko, S.V. Emergence of carbapenemase-producing Gram-negative bacteria in Saint Petersburg, Russia. Int. J. Antimicrob. Agents 2014, 44, 152–155. [Google Scholar] [CrossRef]
- Wyres, K.L.; Wick, R.R.; Judd, L.M.; Froumine, R.; Tokolyi, A.; Gorrie, C.L.; Lam, M.M.C.; Duchene, S.; Jenney, A.; Holt, K.E. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019, 15, e1008114. [Google Scholar] [CrossRef]
- Hamel, M.; Chatzipanagiotou, S.; Hadjadj, L.; Petinaki, E.; Papagianni, S.; Charalampaki, N.; Tsiplakou, S.; Papaioannou, V.; Skarmoutsou, N.; Spiliopoulou, I.; et al. Inactivation of mgrB gene regulator and resistance to colistin is becoming endemic in carbapenem-resistant Klebsiella pneumoniae in Greece: A nationwide study from 2014 to 2017. Int. J. Antimicrob. Agents 2020, 55, 105930. [Google Scholar] [CrossRef]
- Lee, C.R.; Lee, J.H.; Park, K.S.; Jeon, J.H.; Kim, Y.B.; Cha, C.J.; Jeong, B.C.; Lee, S.H. Antimicrobial Resistance of Hypervirulent Klebsiella pneumoniae: Epidemiology, Hypervirulence-Associated Determinants, and Resistance Mechanisms. Front. Cell. Infect. Microbiol. 2017, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Deepbinner: Demultiplexing barcoded Oxford Nanopore reads with deep convolutional neural networks. PLoS Comput. Biol. 2018, 14, e1006583. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garcia-Fernandez, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Moller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample id | Patient Code | Year Collected | Hospital Department | Site | ST | KL-Type | O-Type |
|---|---|---|---|---|---|---|---|
| P-6 | A1 | 2017 | ICU | blood | ST453 | KL8 | O2v2 |
| P-26 | A2 | 2017 | Surgery | traumatic discharge | ST377 | KL102 | O2v2 |
| P-28 | A3 | 2017 | ICU | tracheal aspirate | ST377 | KL102 | O2v2 |
| P-29 | A4 | 2017 | ICU | tracheal aspirate | ST147 | KL10 | O3/O3a |
| P-45 | A5 | 2017 | Urology | Sputum | ST4060* | KL30 | O1v1 |
| P-68 | A6 | 2017 | Rehabilitation | Urine | ST11 | KL24 | O2v1 |
| P-69 | A7 | 2017 | ICU | Sputum | ST29 | KL30 | O1v2 |
| P-75 | A8 | 2017 | ICU | Blood | ST395 | KL108 | O1v2 |
| P-99 | A9 | 2017 | ICU | Urine | ST147 | KL64 | O2v1 |
| P-108 | A10 | 2017 | Thoracic | tracheal aspirate | ST23 | KL57 | O2v2 |
| P-115 | A11 | 2017 | Rehabilitation | Urine | ST35 | KL22 | O1v1 |
| P-116 | A12 | 2017 | ICU | Blood | ST11 | KL27 | O2v2 |
| P-120 | A12 | 2017 | ICU | granulation tissue | ST11 | KL27 | O2v2 |
| P-133 | A13 | 2017 | Rehabilitation | Urine | ST395 | KL2 | O1v1 |
| P-134 | A14 | 2017 | Hematology | Blood | ST395 | KL2 | O1v1 |
| P-137 | A15 | 2017 | Gastroenterology | Urine | ST395 | KL2 | O1v1 |
| P-140 | A16 | 2017 | Hematology | Blood | ST23 | KL57 | O2v2 |
| P-142 | A17 | 2017 | ICU | Faeces | ST147 | KL64 | O2v1 |
| P-152 | A18 | 2017 | Urology | Urine | ST377 | KL102 | O2v2 |
| P-154 | A19 | 2017 | ICU | Blood | ST11 | KL24 | O2v1 |
| P-160 | A20 | 2017 | Hematology | nasal swab | ST307 | KL102 | O2v2 |
| P-176 | A21 | 2017 | ICU | Faeces | ST11 | KL24 | O2v1 |
| P-183 | A22 | 2017 | Cardiology | Urine | ST11 | KL23 | O2v2 |
| P-185 | A23 | 2017 | Rehabilitation | Urine | ST147 | KL64 | O2v1 |
| P-187 | A24 | 2017 | Surgery | traumatic discharge | ST395 | KL2 | O1v1 |
| P-190 | A25 | 2017 | Therapy | Sputum | ST54 | KL14 | O3b |
| P-197 | A26 | 2018 | ICU | Gall bladder | ST26 | KL10 | O1v2 |
| P-200 | A27 | 2018 | Cardiology | Sputum | ST336 | KL25 | O5 |
| P-212 | A28 | 2018 | ICU | bronchoalveolar lavage | ST4061 | KL105 | O4 |
| P-223 | A29 | 2018 | ICU | traumatic discharge | ST395 | KL108 | O1/O2v2 |
| P-226 | A30 | 2018 | Surgery | Blood | ST307 | KL102 | O2v2 |
| P-234 | A31 | 2018 | ICU | abdominal cavity apostem | ST395 | KL39 | O1v1 |
| P-237 | A32 | 2018 | ICU | Blood | ST15 | KL112 | O1v1 |
| P-250 | A33 | 2018 | Hematology | Blood | ST20 | KL111 | O3b |
| P-253 | A34 | 2018 | ICU | Urine | ST15 | KL112 | O1v1 |
| P-259 | A28 | 2018 | ICU | Faeces | ST4061 | KL105 | O4 |
| gapA | infB | mdh | pgi | phoE | rpoB | tonB | ST |
|---|---|---|---|---|---|---|---|
| 2 | 4 | 2 | 1 | 26 | 1 | 2 | 4060 |
| 4 | 1 | 1 | 26 | 66 | 4 | 472 | 4061 |
| Sample id\Plasmid Type | Col | Inc (Other) | IncR |
|---|---|---|---|
| P-6 | Col(BS512) | IncFIB(K)_Kpn3 | - |
| P-26 | ColRNAI | IncHI1B_pNDM-MAR, IncL/M(pOXA-48)_pOXA-48, IncQ1 | - |
| P-28 | ColRNAI | IncHI1B_pNDM-MAR, IncL/M(pOXA-48)_pOXA-48 | - |
| P-29 | Col(BS512) | - | IncR |
| P-45 | - | - | - |
| P-68 | Col(BS512) | IncFIB(K)_Kpn3, IncFII(K) | IncR |
| P-69 | - | - | - |
| P-75 | Col(BS512) | IncFII | IncR |
| P-99 | - | IncFIB(pQil)_pQil, IncHI1B_pNDM-MAR | - |
| P-108 | ColRNAI | IncFII, IncQ1 | - |
| P-115 | - | - | - |
| P-116 | - | IncL/M | IncR |
| P-120 | - | IncL/M | IncR |
| P-133 | - | - | IncR |
| P-134 | - | IncL/M(pOXA-48)_pOXA-48, IncX4 | IncR |
| P-137 | - | IncFIB(Mar)_pNDM-Mar | IncR |
| P-140 | ColRNAI | IncL/M(pOXA-48)_pOXA-48 | - |
| P-142 | - | IncFIB(pQil)_pQil, IncHI1B_pNDM-MAR | - |
| P-152 | ColRNAI | IncHI1B_pNDM-MAR, IncL/M | - |
| P-154 | Col(BS512) | - | IncR |
| P-160 | - | - | - |
| P-176 | Col(BS512) | - | IncR |
| P-183 | ColRNAI | IncA/C2, IncFIB(K)_Kpn3, IncFII(K) | - |
| P-185 | Col440II | IncFII(pKPX1)_AP012055, IncL/M(pOXA-48)_pOXA-48 | IncR |
| P-187 | - | IncL/M | IncR |
| P-190 | - | - | - |
| P-197 | - | - | IncR |
| P-200 | - | IncHI1B_pNDM-MAR | - |
| P-212 | - | IncFII(K) | - |
| P-223 | - | IncL/M(pOXA-48)_pOXA-48 | IncR |
| P-226 | - | IncL/M(pOXA-48)_pOXA-48 | - |
| P-234 | Col(BS512) | IncL/M(pOXA-48)_pOXA-48 | IncR |
| P-237 | Col(BS512) | IncFIB(pQil)_pQil | - |
| P-250 | - | - | - |
| P-253 | Col(BS512) | IncFIB(pQil)_pQil | - |
| P-259 | - | IncFII(K) | - |
| Id | Num_Reads | Num_Contigs | Coverage | N50 |
|---|---|---|---|---|
| P-6 | 9132788 | 347 | 120 | 141817 |
| P-26 | 7655908 | 94 | 87 | 225016 |
| P-28 | 11221516 | 91 | 124 | 249107 |
| P-29 | 8340716 | 121 | 83 | 319263 |
| P-45 | 11426428 | 52 | 192 | 316824 |
| P-68 | 14543708 | 67 | 183 | 261596 |
| P-69 | 15629276 | 57 | 210 | 407523 |
| P-75 | 2399707 | 164 | 64 | 145594 |
| P-99 | 9478824 | 101 | 105 | 206727 |
| P-108 | 7790180 | 82 | 133 | 307643 |
| P-115 | 4387439 | 102 | 120 | 174171 |
| P-116 | 4865862 | 114 | 136 | 159297 |
| P-120 | 4977489 | 155 | 138 | 157758 |
| P-133 | 6947364 | 78 | 125 | 216945 |
| P-134 | 6276120 | 126 | 101 | 160646 |
| P-137 | 7006012 | 103 | 101 | 195276 |
| P-140 | 8749020 | 85 | 144 | 200903 |
| P-142 | 9410400 | 89 | 162 | 277906 |
| P-152 | 577309 | 132 | 28 | 164261 |
| P-154 | 496548 | 92 | 26 | 163095 |
| P-160 | 567724 | 134 | 28 | 203932 |
| P-176 | 17679448 | 78 | 200 | 262658 |
| P-183 | 2441972 | 128 | 50 | 179904 |
| P-185 | 2799220 | 92 | 53 | 230734 |
| P-187 | 3693052 | 139 | 75 | 134707 |
| P-190 | 2891576 | 157 | 59 | 177583 |
| P-197 | 18464812 | 156 | 214 | 145689 |
| P-200 | 22009116 | 114 | 248 | 264170 |
| P-212 | 7357356 | 71 | 155 | 353295 |
| P-223 | 11821000 | 114 | 222 | 260983 |
| P-226 | 9195800 | 107 | 175 | 280687 |
| P-234 | 9399924 | 95 | 155 | 207047 |
| P-237 | 4088672 | 82 | 87 | 209627 |
| P-250 | 18130280 | 97 | 209 | 345165 |
| P-253 | 16252888 | 77 | 173 | 196726 |
| P-259 | 5654368 | 108 | 64 | 241983 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shelenkov, A.; Mikhaylova, Y.; Yanushevich, Y.; Samoilov, A.; Petrova, L.; Fomina, V.; Gusarov, V.; Zamyatin, M.; Shagin, D.; Akimkin, V. Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates. Antibiotics 2020, 9, 261. https://doi.org/10.3390/antibiotics9050261
Shelenkov A, Mikhaylova Y, Yanushevich Y, Samoilov A, Petrova L, Fomina V, Gusarov V, Zamyatin M, Shagin D, Akimkin V. Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates. Antibiotics. 2020; 9(5):261. https://doi.org/10.3390/antibiotics9050261
Chicago/Turabian StyleShelenkov, Andrey, Yulia Mikhaylova, Yuri Yanushevich, Andrei Samoilov, Lyudmila Petrova, Valeria Fomina, Vitaly Gusarov, Mikhail Zamyatin, Dmitriy Shagin, and Vasiliy Akimkin. 2020. "Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates" Antibiotics 9, no. 5: 261. https://doi.org/10.3390/antibiotics9050261
APA StyleShelenkov, A., Mikhaylova, Y., Yanushevich, Y., Samoilov, A., Petrova, L., Fomina, V., Gusarov, V., Zamyatin, M., Shagin, D., & Akimkin, V. (2020). Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates. Antibiotics, 9(5), 261. https://doi.org/10.3390/antibiotics9050261