1. Introduction
Antimicrobial resistance (AMR) genes are inherently present in bacteria. However, the (mis)use of antibiotics increased the global prevalence of AMR; as a result, it has become one of the most prominent public health threats [
1]. Plasmids are mobile genetic elements that can carry various genes beneficial for the survival of bacteria, including AMR genes. Plasmids are highly diverse in their genetic content, size (1–1000 kb) and copy number (1−500). Bacteria can exchange plasmids between isolates even across the species or phyla barriers, also called horizontal gene transfer (HGT) [
2,
3,
4,
5,
6]. This transfer of plasmids facilitates the transmission of accompanying AMR genes, making plasmid detection and reconstruction of the utmost importance to understand the dissemination of AMR genes and to improve risk assessment.
Due to the globalization of the modern world, plasmids with AMR genes of clinical relevance can disseminate in a short time span [
3,
7,
8]. There has been an increase of extended-spectrum β-lactamase (ESBL)-producing Enterobacterales (containing variants of the
blaTEM,
blaSHV and
blaCTX-M ESBL genes) reported in Europe caused by specific clones and plasmids [
9]. Moreover, the carbapenemase genes encoding for OXA-48, KPC, VIM, NDM and IMP have been increasingly detected, and while carbapenemase-producing Enterobacterales (CPE) used to only be reported in hospital settings, nowadays CPE are also detected in community and environmental samples in Europe [
10]. Moreover, these carbapenemase genes have also been increasingly reported on plasmids [
11]. Of extra concern is that the described plasmids are often carrying multiple AMR genes, making treatment of infections caused by bacteria carrying these plasmids even more troublesome and sometimes leaving only colistin as a last resort antimicrobial. Unfortunately, the
mcr-1 gene conferring resistance to the last-line antibiotic colistin has recently been detected on a plasmid in China [
12]. Shortly afterwards,
mcr-1 was detected worldwide with co-localization of ESBL genes on a plasmid [
13,
14,
15,
16]. Furthermore, the European Food Safety Authority (EFSA) reported that food-borne infections are becoming harder to treat as a consequence of the increase in AMR prevalence [
17]. However, the data on circulating plasmids are only slowly increasing, and in particular, detailed knowledge on the plasmid structure and rearrangements is severely lacking, which can hinder risk assessment [
18].
There are several techniques to detect AMR genes and plasmids, of which PCR is a commonly used one [
19,
20,
21,
22,
23]. (q)PCR allows to indicate the presence of a plasmid or AMR gene, with one targeted PCR test per AMR gene/plasmid locus to be identified [
20,
21]. However, the determination whether the AMR gene is localized on a plasmid necessitates additional experiments including time-consuming Southern blotting [
24] or plasmid transfer followed by further analyses [
25]. With next-generation sequencing (NGS), multiple aspects such as the complete resistance profile, including the AMR gene sequence, and plasmid elements can be determined simultaneously, without a priori knowledge [
22,
23]. Short-read sequencing technologies showed to have high accuracy in detecting the AMR genes, with some reports stating high concordance with phenotypic results in Enterobacterales [
26]. However, plasmids often contain large repetitive regions that cannot be bridged by short sequencing reads [
27,
28,
29,
30], resulting in incomplete contigs of which the origin, chromosome or plasmid, is uncertain. If the AMR genes are localized on these incomplete contigs, it is uncertain whether they are localized on a plasmid (higher risk of transfer). Similarly, there are insertion sequences (IS) of 600–7900 bp which contain AMR genes and are flanked by short direct repeats [
31]. IS can be present on either the chromosome or a plasmid; however, their location cannot always be resolved with short-read sequencing. Since the introduction of third-generation sequencers (Oxford Nanopore Technologies and Pacific Biosciences (PacBio)), it is possible to generate long sequencing reads (>1000 bp) in high throughput that are able to bridge these repetitive regions [
30,
32,
33]. However, these long reads also contain more errors (10–20% error rate) compared to short reads, which can hinder AMR gene prediction. Others have shown that by using short reads in combination with long reads, it is possible to reconstruct full genomes, including plasmids [
30,
33,
34,
35,
36]. Aside from the higher cost of sequencing with two technologies, another issue is that long-read sequencing requires high amounts of input DNA of high molecular weight [
37]. These restrictions might not always be compatible with the current DNA extraction protocols routinely used for surveillance. Therefore, to obtain the most optimal plasmid reconstruction with NGS, there are multiple parameters to consider, both at the wet and dry lab level. However, specific guidelines on what would be the best approach to take for efficient plasmid reconstruction are still missing.
One parameter to consider is the input DNA material to be used. As a starting isolate, the plasmid-containing original strain could be used, or the plasmid could first be transferred to a lab-host recipient with a known chromosome sequence [
38]. Although a transfer would take more hands-on time compared to working directly with the original (clinical, food, environmental) isolates, the benefit would be that as the complete chromosome sequence is already known, reads matching to this sequence could be filtered out during the bioinformatics analysis, in theory only leaving plasmid reads in the data set for plasmid reconstruction [
22]. Alternatively to this in silico approach to retrieve plasmid reads, a plasmid DNA extraction method could be used instead of a method targeting the total genome [
39,
40], thereby leaving only plasmid DNA as input for the NGS approach. Most plasmid DNA extractions are based on the difference in size between chromosomal and plasmid DNA and start with an alkaline lysis [
41] that denatures all DNA. Shorter DNA fragments (plasmids) renature more quickly, and this enables them to be separated from the chromosomal DNA by several methods [
42]. However, this is not always successful, as some plasmids might be lost during extraction, or chromosomal DNA may remain in the final extract. It has been reported that column-based plasmid DNA extractions have difficulties with extracting plasmids >150 kb in size [
43]. Additionally, an insufficient amount of plasmid DNA might be obtained [
44,
45], which, as mentioned, might be an issue for long-read technologies (Nanopore or PacBio) which require >500 ng of high-molecular-weight input DNA depending on the library preparation protocol [
37,
46]. Nevertheless, plasmid DNA extractions have been successfully used with long-read sequencing for the reconstruction of plasmids [
33,
36]. However, a systematic comparison on the effect of different DNA extraction protocols on plasmid reconstruction has not yet been reported.
Another parameter to consider is the sequencing technology. For short-read sequencing, Illumina has been by far the most used. However, as elaborated above, short reads only might be insufficient for plasmid reconstruction. There are currently two accessible long-read sequencing technologies, i.e., from Nanopore and PacBio. While the error rate of the PacBio technology is lower [
47], Nanopore technology produces a higher yield and longer reads [
48]. Moreover, an important advantage of the MinION flow cells from Nanopore is that they are accessible to more labs due to the lower initial investment and size [
47]. Additionally, the MinION flow cells are more cost-efficient, which is important to consider when a large number of isolates in routine analyses or surveillance need to be sequenced. Recently, Nanopore released the Flongle, which is a smaller version of the MinION, making it ideal for sequencing a single bacterial isolate. However, the Flongle has not yet been tested for the reconstruction of plasmids.
The last parameter to consider is the bioinformatic analysis. After sequencing, the reads are usually trimmed and filtered [
49]. Then, the overlap between sequencing reads (assembly) is used to reconstruct the genomic structures, or the reads are directly used in alignments to known plasmid sequences. The overlap between sequencing reads is determined automatically by software (assembly tools) that makes use of only short or long reads, or a combination of both sequencing reads (hybrid). As aforementioned, using only short-read sequencing for reconstruction of plasmids is often insufficient due to the repetitive regions in plasmids [
27,
28,
29,
30], and therefore long-read sequencing is needed. In routine testing, however, cost and accuracy of AMR detection and full plasmid reconstruction have to be taken into account. Nevertheless, it has not been systematically evaluated yet whether long-read only or hybrid assemblies are more suitable for this purpose. Subsequently, more information can be retrieved from the assemblies by comparing them to databases for AMR genes [
50,
51] or plasmid replicons/sequences [
52,
53,
54].
Although there have been other studies that compared different (plasmid) DNA extraction methods and/or assemblies [
44,
55,
56,
57,
58,
59], these studies only focused on one of the aforementioned parameters. Moreover, for some of these studies, the plasmid reconstruction was not the main focus, and therefore the completeness or AMR profile of the reconstructed plasmid(s) was not evaluated. As the setup/focus of these studies was very different, involving, e.g., different isolates and plasmids, it is difficult to compare the results in view of a systematic evaluation. Therefore, this study aimed to determine the effect of the above-described parameters on the NGS results and the reconstruction of plasmids in the context of AMR monitoring. In view of proposing an optimal workflow covering both the wet and dry lab, the reconstruction of an
mcr-1 plasmid from several whole-genome and plasmid DNA extracts with only MiSeq or MinION data and a combination of both (hybrid) were compared. Both the original isolate and the corresponding transconjugant were used. Furthermore, the newly released Flongle flow cell from Nanopore was used for sequencing, and the produced data were evaluated for the feasibility of plasmid reconstruction. Finally, the obtained workflow was applied to multidrug-resistant
Salmonella Kentucky isolates for which it had been impossible to determine with solely short-read sequencing whether the clinically relevant ESBL genes were localized on a plasmid or on the chromosome.
3. Discussion
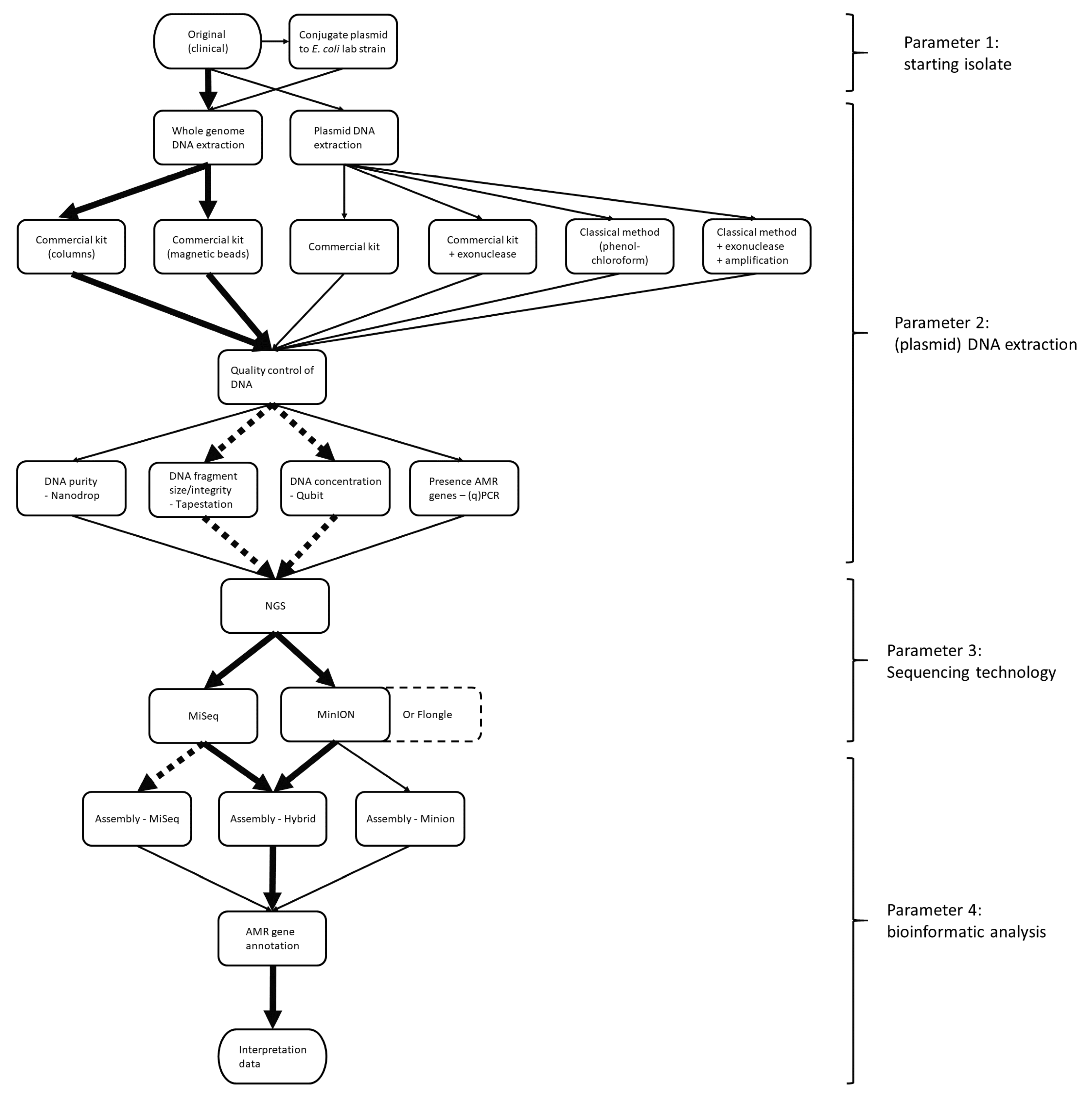
AMR constitutes a huge public health concern. As plasmids contribute to the spread of AMR genes, their occurrence should be monitored. NGS is a powerful tool for the surveillance of AMR. However, it is difficult to reconstruct plasmids based on the commonly used short-read NGS because of the repetitive structures in plasmids. Therefore, it is challenging to determine whether AMR genes are located on the chromosome or on a plasmid within an isolate, which also hinders proper risk assessment and monitoring of circulating plasmids. In the current study, a strategy to reconstruct plasmids using NGS data in the context of AMR gene surveillance was determined. For this, several parameters at different levels of the workflow in both the wet and dry lab were evaluated (
Figure 1). Within the workflow, the most optimal starting isolate (parameter 1), DNA extraction (parameter 2), sequencing technology (parameter 3) and bioinformatic analyses (parameter 4) were identified, using
mcr-1-containing plasmids as case studies.
In a first attempt to reduce the complexity of plasmid reconstruction based on NGS data, a conjugation was performed in which a plasmid with unknown sequence was transferred to a lab host for which the genome sequence was known. In theory, this would facilitate the computational analyses, as the chromosomal reads could be filtered out in silico by mapping to the known genome sequence of the host strain, leaving only the plasmid reads to be used in assembly and hence for plasmid reconstruction. With only MiSeq reads, this filtering strategy did provide the AMR profile of the plasmid; however, the assembly was still very fragmented. Nevertheless, it was less fragmented than the MiSeq assembly without any chromosomal filtering step. With only MinION reads, the general structure of the plasmid could be retrieved; however, the size was incorrect, AMR genes had lower sequence identity to the ResFinder database than the MiSeq and hybrid assemblies and it was not possible to circularize the contig. A combination of the filtered MiSeq and MinION reads resulted in an almost complete reconstruction. However, it was missing a small fragment of ~1000 bp that it shared with the chromosome of the lab
E. coli, and therefore any plasmid reads that contained this sequence were filtered out. The hybrid assembly of the conjugated sample without any filtering resulted in a complete chromosome and plasmid. Nevertheless, with whole-genome sequencing of conjugated isolates, a large part of the sequencing capacity is wasted, as the chromosomal sequence is already known. Moreover, as described in this study, it is possible for isolates to contain multiple plasmids, which can cause problems with the conjugation. Some plasmids will be nonconjugative (i.e., unable to transfer to another isolate or species) [
2], or there will not be enough selection pressure if only antibiotics are administered of which the AMR genes are localized on other plasmids. However, as shown in this study, these nonconjugative plasmids can contain clinically relevant AMR genes, and these are unable to be characterized in the conjugated samples. Alternatively, transformation/electroporation could be considered. On the other hand, by transferring only one plasmid, it allows for the phenotypic effects to be studied with less interference of other genes, which would allow showing that the observed phenotype is the sole consequence of the presence of that particular plasmid. Because of the disadvantages combined with the fact that conjugation experiments take more hands-on time, it was decided that for the reconstruction of plasmids with hybrid assemblies it is more beneficial to start with the original isolate. However, it was seen that with the MiSeq reads of the conjugated samples where the chromosomal sequences were filtered out, it was possible to determine whether an AMR gene was located on the chromosome or a plasmid without full plasmid reconstruction. Therefore, if no long-read sequencing can be performed, it can be of added value to analyze conjugated samples with short-read sequencing only to determine the AMR profile of the transferred plasmid.
An alternative approach to reduce the chromosomal reads in the final output is to limit the input for the NGS analysis to plasmid DNA by adapting the DNA extraction protocol. As the goal is to use the workflow within a surveillance system in place in a National Reference Centre, it is important that the extraction method is cost-efficient with a limited hands-on time. Not all extraction methods are equally suited for the recovery of (large) plasmids. Moreover, long-read sequencing requires high-quality DNA, in large quantities and of high molecular weight [
37]. Taking these restrictions into account, several plasmid extraction protocols were evaluated and compared to two whole-genome DNA extraction methods. This comparison was based on the quality parameters of the DNA and reconstruction of the plasmid with short (MiSeq) and long (MinION) sequencing reads and a combination of both. The plasmid DNA extractions were of much lower concentration than the whole-genome DNA extractions, even if an amplification step was performed. In the assemblies, plasmid DNA extractions contained many small incomplete contigs; without prior knowledge about the plasmid profile of the isolate, it would be difficult to determine whether these are of chromosomal or plasmid origin. Cloning vectors and naturally occurring plasmids can contain regions originating from chromosomes [
65] or chromosomes can integrate plasmid sequences by homologous recombination [
66], and therefore it becomes difficult to determine if these unexpected linear contigs are part of chromosomes or plasmids. The only plasmid DNA extraction which did not contain incomplete contigs was the phenol chloroform combined with an exonuclease and amplification of the DNA. However, the lower concentration still caused problems in the MinION sequencing, making the hybrid assembly of lower quality than the whole-genome DNA extractions. Moreover, this plasmid extraction protocol is more laborious and expensive than the whole-genome extraction. Although the proportion of reads mapping to the plasmid was much lower in whole-genome extracts (~5% vs. >50%), these reads were still sufficient for fully reconstructing the plasmid. Furthermore, with the whole-genome DNA extractions, the copy number of the plasmid could also be estimated by comparing the plasmid coverage to that of the chromosome. Although the tested plasmid DNA extractions were able to reconstruct the large, low-copy
mcr-1 plasmid, the whole-genome DNA extractions performed equally as well or better in all tested parameters. Therefore, the whole-genome DNA extractions were chosen for the workflow. However, if only short-read sequencing can be performed and it concerns a crucial isolate for which a specific, more time-consuming DNA extraction protocol can be followed, then the phenol chloroform plasmid DNA extraction with exonuclease and amplification can be useful to determine the plasmid content.
The most reliable reconstruction of plasmids was with hybrid assemblies from the whole-genome Genomic Tip 100 and MagCore DNA extracts of the original isolate. A benefit for AMR surveillance of reconstructing plasmids with whole-genome assemblies is that simultaneously chromosomal and plasmid AMR genes can be determined. While the Genomic Tip 100 is more expensive and more time-consuming per sample than the automated MagCore DNA extractions, it also produced more consistently longer Nanopore reads. However, the smaller MinION reads from the MagCore extract were still sufficient to cover all repetitive regions in the hybrid assembly of the used case study. Therefore, both (or similar whole-genome DNA extractions) can be used in the workflow. However, even though the MagCore showed lower DNA purities and inconsistent fragment sizes, it is more adapted to a routine surveillance setting than the Genomic Tip 100 extractions as the MagCore extractions are more cost-efficient and require less hands-on time (semiautomatization). Based on the DNA quality tests, it seemed that concentration and fragment sizes had a larger effect on the Nanopore read length and yield than the purity of the DNA extract. The contaminants that caused the lower purity in the MagCore extracts are likely removed during the cleaning steps with the magnetic AMPure XP beads in the library preparation of the 1D ligation sequencing kit.
In this study, Unicycler was used for all assemblies to keep the workflow consistent. Unicycler is a pipeline consisting of several tools that perform the assembly, error correction and circularization of contigs. It has been shown to produce very accurate hybrid assemblies [
67]. Another advantage of Unicycler is that it is relatively user-friendly, requiring a single command on the command line. Although Unicycler was used in a Linux environment, which might be more difficult to put into place in a routine surveillance setting, there are currently no hybrid assemblers that are usable in Windows. However, a user-friendly GUI is available for nonexperts [
68]. Nevertheless, there are other (hybrid) assemblers available that specifically focus on plasmids (e.g., plasmidSPAdes) [
69] or do reference-guided assemblies (e.g., PLACNETw) [
70] or that focus just like Unicycler on the whole genome including plasmids (e.g., Canu) [
71]. However, plasmidSPAdes has been shown to have difficulties with assembling large and low-copy plasmids [
27]. Furthermore, reference-guided assemblers such as PLACNETw could misassemble plasmids that contain rearrangements and plasmids that are not present in the database or of which there are only low-quality references in the database. It would be interesting to make an in-depth comparison between all available software in a dedicated future study.
Based on the above, it was seen that a hybrid assembly from a whole-genome extraction of the original isolate was the best approach for accurate plasmid reconstruction using NGS data. Next, it was determined which sequencing technology and assembly method resulted in the most accurate AMR gene prediction and localization. The similarity of the detected genes to the ones in the AMR databases used (percent identity) was higher in MiSeq assemblies than in the Nanopore assemblies, which can be explained by the lower error rate of short-read sequencing. With the lower similarity, it was, however, still possible to determine the correct variant in the Nanopore assemblies. Nevertheless, care should be taken, as for gene variants that only differ by a few base pairs (for example blaCTX-M), this could result in the wrong gene prediction. Moreover, the detection of the correct gene or variant is also dependent on the content of the database and alignment software. It would be helpful for AMR gene databases to be harmonized and kept continuously updated in the future. Furthermore, the Nanopore assembly is not suitable for the prediction of chromosomal mutations (SNPs) that confer AMR. As to the localization of the AMR genes, in MiSeq assemblies it was impossible to determine whether the AMR gene was located on a chromosome or plasmid, in contrast to the MinION assemblies. Therefore, hybrid assemblies combined the best of both worlds, i.e., resulting in both very accurate AMR gene prediction and correct localization, making them the most suitable for the workflow.
As hybrid assemblies gave the most complete assemblies of plasmids with accurate AMR gene prediction, both short and long sequencing reads would be necessary. In this study, Nanopore sequencing was chosen, as this portable technology is accessible to more labs and has a lower initial investment cost compared to PacBio. Nanopore technologies offer two types of flow cells, i.e., the already widely used MinION and the newer Flongle. Although the Flongle is cheaper, it has a lower pore count and therefore produces less output. However, this makes this flow cell interesting for analyzing a single sample at a time, thereby avoiding the need for barcoding of samples when using the MinION flow cell, which requires the grouping of ~12 samples to be cost-effective. While the MinION can be used to save time if multiple samples need to be analyzed simultaneously, between 1–5% of the reads are lost because the barcode cannot be recognized. Another disadvantage of barcoding is the occurrence of cross-over contamination between barcodes, which is mostly caused by chimeric reads [
72]. While this contamination has been shown to not affect hybrid assemblies [
57], it could result in erroneous alignments when the sequencing reads are used for this purpose. The use of a Flongle will avoid this issue and will also allow for a more rapid turn-around time of a hybrid assembly analysis in a daily routine setting, as there is no need to wait until a sufficient number of samples are collected to start a MinION analysis. Additionally, the Flongle requires less input DNA per sample, i.e., 500 ng compared to 1 µg for MinION flow cells. For these reasons, we compared the sequencing output and hybrid assemblies from the MinION to the Flongle and determined that the Flongle output is more than sufficient to reconstruct plasmids and also the bacterial chromosome. Moreover, the AMR prediction accuracy of the hybrid and Nanopore-only assemblies from Flongle data was similar to that of assemblies with MinION sequencing reads. Therefore, the Flongle seems more suitable for the workflow, except when a lot of isolates need to be sequenced in parallel.
One issue with Nanopore sequencing, in contrast to the routinely used short-read sequencing technology, is that the protocols for library preparation as recommended by Oxford Nanopore Technologies are constantly evolving. When starting this study, it was recommended to fragment the input DNA to 8 kb during the library preparation with the 1D ligation sequencing kit SQK-LSK108 to increase the sequencing output. Since the introduction of the ligation sequencing SQK-LSK109 kit, shearing of input DNA was only recommended when using amounts of DNA which are below the specifications of Oxford Nanopore Technologies. Moreover, the 1D ligation sequencing kit was chosen for its higher sequencing output and reliability at the start of the study. Another library preparation kit that has been used on plasmids is the rapid sequencing kit (SQK-RAD). With the rapid sequencing kit, less manipulations are required in the library preparation, which may be useful in obtaining longer reads or even ultralong reads (>100 kb) [
33,
73,
74]. However, the rapid sequencing kit does not have any cleaning steps during the library preparation, and therefore a larger effect of the purity of the DNA on the sequencing results might be seen than with the 1D ligation sequencing kit. In this study, different library preparations were compared to determine the effect of DNA fragmentation on sequencing output, read length and plasmid reconstruction. The results showed that fragmentation increased the obtained sequencing output; however, this was at the expense of the average read size, which was smaller compared to that obtained with unfragmented samples. However, both with and without fragmentation, the plasmid was completely reconstructed with high accuracy for all expected AMR genes. While the read size did not matter for the assembly of our specific case study, it is advisable not to shear the DNA of the isolate if the DNA concentration is within the recommendations of Oxford Nanopore Technologies (>500 ng for Flongle and >1000 ng for MinION) to ensure that all repetitive regions can be bridged.
Performing both short- and long-read sequencing for each isolate can be costly, especially in a surveillance context where many isolates need to be analyzed. Therefore, it would be recommended to first perform short-read sequencing on all isolates to determine if there are indications of unique or otherwise relevant plasmids or clinically relevant AMR genes, and then select those for additional long-read sequencing. While with MiSeq sequencing only it was not possible to accurately reconstruct plasmids, it is possible to get an indication as to whether there were sequences that likely come from plasmids using databases like PlasmidFinder [
52] and PLSDB [
53] or machine-learning tools [
75]. However, there are still improvements possible in these databases and tools as, for example, some plasmids contain multiple or zero of the replicons described in the PlasmidFinder database [
52,
76], and machine learning is highly dependent on the training dataset and comes with its own biases and pitfalls [
77,
78,
79]. In the future, it is likely that the accuracy and output of long-read sequencing keeps improving, and thus it would then be possible to replace hybrid assemblies with assemblies made solely from long-read sequences if accurate AMR gene detection and localization is of interest. The R10 flow cells are reported to have higher accuracy. It would be interesting to test whether the accuracy is sufficient to be able to reduce our workflow to long-read sequencing only.
Our optimal workflow for plasmid reconstruction, consisting of whole-genome DNA extraction on the original isolate followed by MiSeq and Nanopore sequencing to perform hybrid assemblies, was established using isolates with a large
mcr-1-containing plasmid. Without application of this workflow, it would not have been possible to reconstruct this large, low-copy plasmid of >200 kb. Besides the
mcr-1 gene, the plasmid contained ESBL genes and other critically important AMR genes [
80]. We applied the workflow both to
E. coli and
Salmonella. The NCBI nucleotide database contained sequences similar to that of the
mcr-1 plasmid from the
E. coli isolates (COL20160015 and R274) but did not contain a sequence that completely covered the
mcr-1 plasmid from the
Salmonella isolate (S15BD05371). This highlights that the public databases are currently lacking sequences of circulating plasmids. To be able to trace these plasmids and prevent their spread, a more complete characterization of plasmids and submission of these sequences to public repositories are needed.
Finally, our workflow was applied to an ESBL-positive
Salmonella Kentucky case study. ESBL-positive
S. Kentucky are part of the list of multidrug-resistant pathogens that have been designated as a high-priority issue by the WHO [
81]. In the beginning of the 21st century, European ST198
Salmonella Kentucky were susceptible to antibiotics; however, there was a rapid increase of mutations in the
parC and
gyrA genes that are involved in ciprofloxacin resistance. Of extra concern is the recent acquisition of bla
CTX-M-14b gene reported in ST198
S. Kentucky that was not seen before and had already started to spread throughout Europe [
61,
82]. At the start of this study, it was unknown whether the ESBL AMR genes detected by short-read sequencing were localized on a plasmid or on the chromosome. Without this knowledge, it is more difficult to assess the risk of spread of this AMR, because while chromosomal ESBL genes spread clonally within the
S. Kentucky population, it has the potential to spread to other bacterial pathogens if it is localized on a plasmid. With the hybrid assemblies obtained with the workflow, it was determined that in the Belgian
S. Kentucky isolates, the ESBL genes were present on both the chromosome and on plasmids and that there was also a likely exchange by a transposase of a 2850 bp region between the chromosome and a plasmid of different isolates. The results of our workflow on the Belgian isolates, and especially of the plasmid reconstruction, were put into epidemiological context of the other European
Salmonella Kentucky ESBL-positive isolates by Coipan et al. [
83]. They found that this 2850 bp fragment was present in the chromosome and plasmids of other
S. Kentucky isolates [
83]. The exchanged fragment is very similar to that on the chromosome of an
S. Kentucky isolated from poultry in China [
64]; however, the exact sequence reported in the Chinese study is not yet available in public databases to make the full comparison. Other surveillance and in vitro studies have also shown the role of this transposase in transfer of
blaCTX-M genes from plasmids to chromosomes of bacteria [
84]. Due to the lack of data in current databases, it could not be determined whether the 2850 bp fragment was first localized on the chromosome and then transferred to the detected plasmid or vice versa. However, the presence of the ESBL gene on the plasmid increases its transmissibility, which is a public health risk. Without the application of our developed workflow, this knowledge would not have been available.
This study describes a systematic evaluation of wet and dry lab components involved in accurate reconstruction of plasmids and characterization of their AMR gene content based on NGS data. Our developed workflow will contribute to an improved monitoring of circulating plasmids and assessment of their transfer risk.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}