
Hierarchical Porous Carbon Fibers Synthesized by Solution-Plasma-Generated Soot Deposition and Their CO2 Adsorption Capacity

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. The Solution Plasma (SP) Reactor and Heating Chamber
2.2. Synthesis of Hierarchical Carbon Fibers (HCF)
2.3. HCF Post Treatment
2.4. Characterization
3. Results
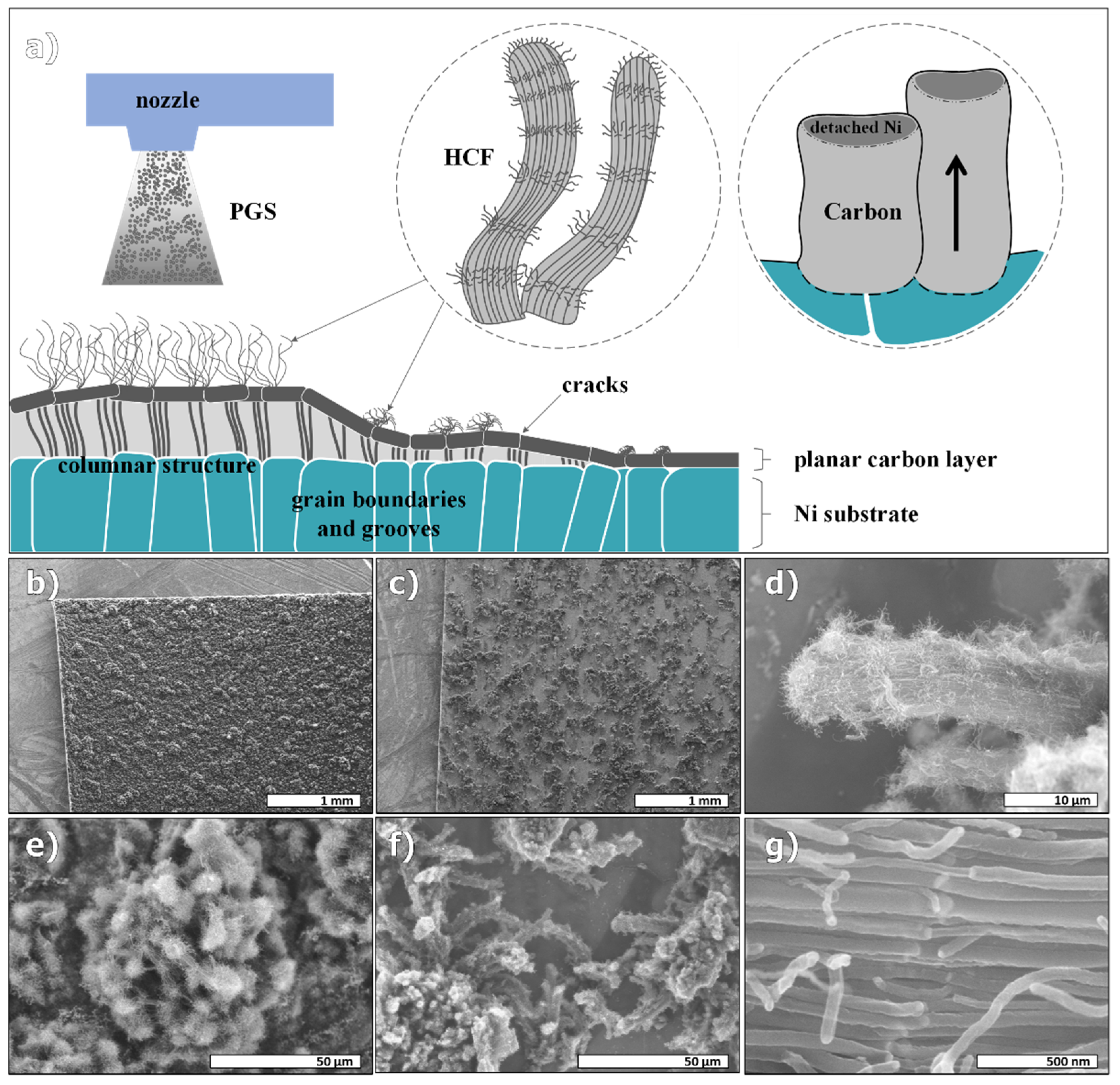
3.1. Hierarchical Structure
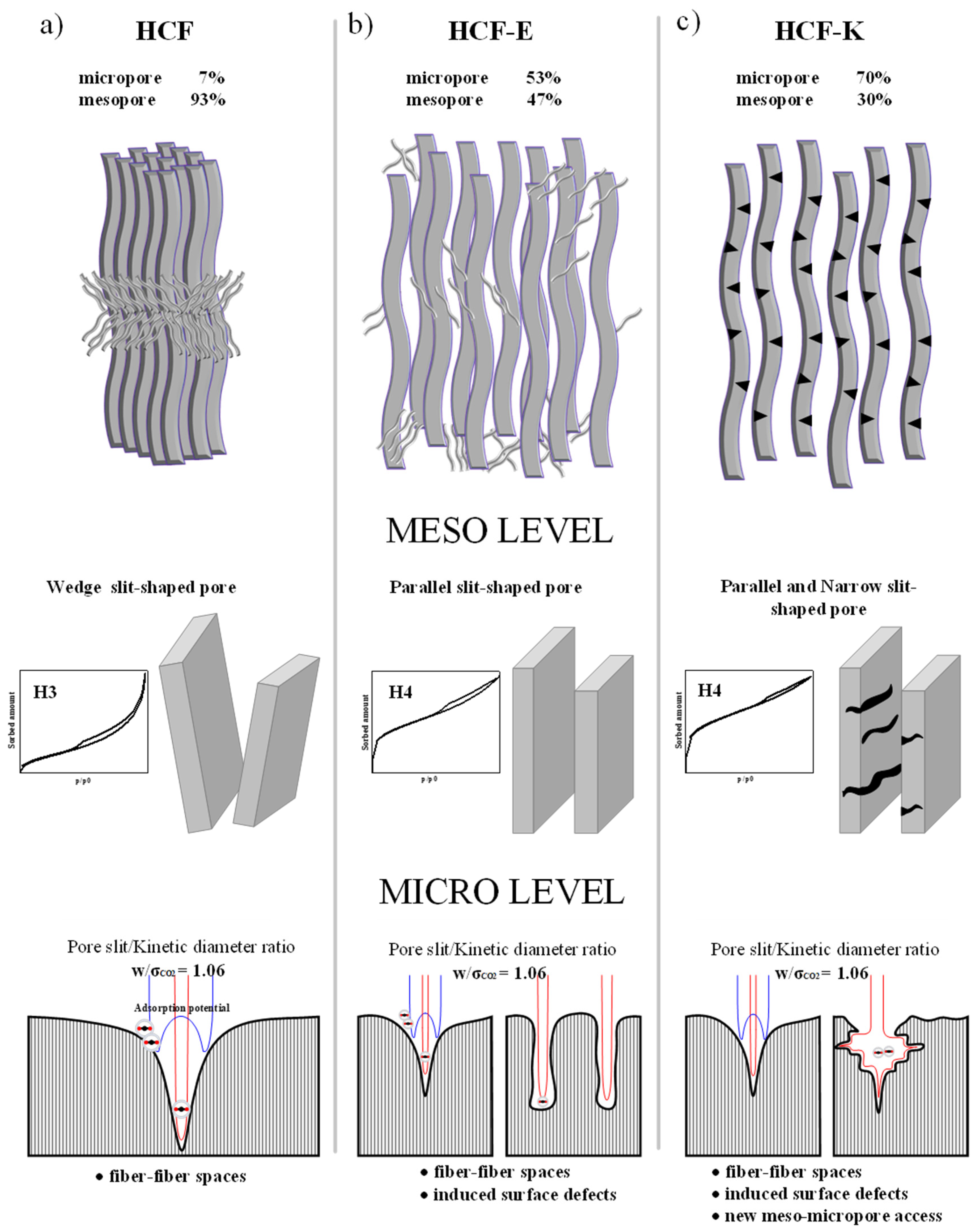
3.2. Post Modification of HCFs
3.3. CO2 Adsorption of HCFs
4. Discussion
4.1. Hierarchical Structure
4.2. Morphology and CO2 Adsorption Synergy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manabe, S. Modeling Study of Greenhouse Warming—A Citation-Classic Commentary on Thermal-Equilibrium of the Atmosphere with a Given Distribution of Relative-Humidity by Manabe, S. and Wetherald, R.T. Cc/Eng. Technol. Appl. Sci. 1993, 8, 8. [Google Scholar]
- Yaghi, O.M. Reticular Chemistry in All Dimensions. ACS Central Sci. 2019, 5, 1295–1300. [Google Scholar] [CrossRef] [Green Version]
- Lyu, H.; Li, H.; Hanikel, N.; Wang, K.; Yaghi, O.M. Covalent Organic Frameworks for Carbon Dioxide Capture from Air. J. Am. Chem. Soc. 2022, 144, 12989–12995. [Google Scholar] [CrossRef]
- Li, J.; Zhao, D.; Liu, J.; Liu, A.; Ma, D. Covalent Organic Frameworks: A Promising Materials Platform for Photocatalytic CO2 Reductions. Molecules 2020, 25, 2425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Dong, B.; Ge, R.; Wang, C.; Song, X.; Ma, W.; Wang, Y.; Hao, C.; Guo, X.; Gao, Y. Channel-wall functionalization in covalent organic frameworks for the enhancement of CO2 uptake and CO2/N2 selectivity. RSC Adv. 2016, 6, 38774–38781. [Google Scholar] [CrossRef]
- Kuroda, K. Discovery of mesoporous silica from layered silicates. In Studies in Surface Science and Catalysis; Terasaki, O., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 148, pp. 73–108. [Google Scholar]
- Lee, J.-S.M.; Cooper, A.I. Advances in Conjugated Microporous Polymers. Chem. Rev. 2020, 120, 2171–2214. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, M. Chapter 6—Intercalation compounds New Carbons—Control of Structure and Functions; Inagaki, M., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2000; pp. 146–176. [Google Scholar]
- Inagaki, M. Chapter 5—Porous carbons New Carbons—Control of Structure and Functions; Inagaki, M., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2000; pp. 124–145. [Google Scholar]
- Siegelman, R.L.; Kim, E.J.; Long, J.R. Porous materials for carbon dioxide separations. Nat. Mater. 2021, 20, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Li, J.; Liu, A.; Chen, C. Carbon Gels-Modified TiO2: Promising Materials for Photocatalysis Applications. Materials 2020, 13, 1734. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Li, Y.; Cao, M.; Hu, C. A novel activating strategy to achieve highly porous carbon monoliths for CO2 capture. J. Mater. Chem. A 2014, 2, 4819–4826. [Google Scholar] [CrossRef]
- Srinivas, G.; Krungleviciute, V.; Guo, Z.-X.; Yildirim, T. Exceptional CO2 capture in a hierarchically porous carbon with simultaneous high surface area and pore volume. Energy Environ. Sci. 2013, 7, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Zhang, H.; Sun, H.; Suvorova, A.; Saunders, M.; Tade, M.; Wang, S. Heteroatom (N or N-S)-Doping Induced Layered and Honeycomb Microstructures of Porous Carbons for CO Capture and Energy Applications. Adv. Funct. Mater. 2016, 26, 8651–8661. [Google Scholar] [CrossRef]
- Singh, S.K.; Akhtar, M.J.; Kar, K.K. Hierarchical Carbon Nanotube-Coated Carbon Fiber: Ultra Lightweight, Thin, and Highly Efficient Microwave Absorber. ACS Appl. Mater. Interfaces 2018, 10, 24816–24828. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, C.; Zhou, Y.; Zhao, W.; Zhong, J.; Huang, W.; Chen, R. Fabrication of Hierarchical N-doped Carbon Nanotubes for CO2 Adsorption. Nano 2019, 14, 1950072. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, T.; Khan, A.U.; Liu, G. Block copolymer–based porous carbon fibers. Sci. Adv. 2019, 5, eaau6852. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.W.; White, J.L.; Buechler, M. Mesophase/isotropic phase interfacial energy: Determination from coalescence kinetics of mesophase pitch. Carbon 1985, 23, 117–121. [Google Scholar] [CrossRef]
- Endo, M.; Koyama, T.; Hishiyama, Y. Structural Improvement of Carbon Fibers Prepared from Benzene. Jpn. J. Appl. Phys. 1976, 15, 2073–2076. [Google Scholar] [CrossRef]
- Oberlin, A.; Endo, M.; Koyama, T. Filamentous growth of carbon through benzene decomposition. J. Cryst. Growth 1976, 32, 335–349. [Google Scholar] [CrossRef]
- Endo, M.; Kroto, H.W. Formation of carbon nanofibers. J. Phys. Chem. 1992, 96, 6941–6944. [Google Scholar] [CrossRef]
- Inagaki, M. Chapter 4—Carbon Fibers New Carbons—Control of Structure and Functions; Inagaki, M., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2000; pp. 82–123. [Google Scholar]
- Kang, J.; Li, O.L.; Saito, N. Synthesis of structure-controlled carbon nano spheres by solution plasma process. Carbon 2013, 60, 292–298. [Google Scholar] [CrossRef]
- Hyun, K.; Saito, N. The solution plasma process for heteroatom-carbon nanosheets: The role of precursors. Sci. Rep. 2017, 7, 3825. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.; Panomsuwan, G.; Bratescu, M.A.; Teshima, K.; Saito, N. p-Type Doping of Graphene with Cationic Nitrogen. ACS Appl. Nano Mater. 2019, 2, 1350–1355. [Google Scholar] [CrossRef]
- Kim, K.; Chokradjaroen, C.; Saito, N. Solution plasma: New synthesis method of N-doped carbon dots as ultra-sensitive fluorescence detector for 2,4,6-trinitrophenol. Nano Express 2020, 1, 020043. [Google Scholar] [CrossRef]
- Saito, N.; Hieda, J.; Takai, O. Synthesis process of gold nanoparticles in solution plasma. Thin Solid Films 2009, 518, 912–917. [Google Scholar] [CrossRef]
- Morishita, T.; Ueno, T.; Panomsuwan, G.; Hieda, J.; Yoshida, A.; Bratescu, M.A.; Saito, N. Fastest Formation Routes of Nanocarbons in Solution Plasma Processes. Sci. Rep. 2016, 6, 36880. [Google Scholar] [CrossRef] [Green Version]
- Pakhnevich, A.A.; Golod, S.V.; Prinz, V.Y. Surface melting of copper during graphene growth by chemical vapour deposition. J. Phys. D Appl. Phys. 2015, 48, 435303. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Carbon Deposition in Steam Reforming and Methanation. Catal. Rev. 1982, 24, 67–112. [Google Scholar] [CrossRef]
- Hayashi, T.; Karita, M.; Nakano, T.; Inoue, Y. A study on the growth enhancement effects of chlorine on carbon nanotube forest in chloride-mediated chemical vapor deposition. Jpn. J. Appl. Phys. 2021, 60, 045001. [Google Scholar] [CrossRef]
- Schmidt, I.; Benndorf, C. Using fluorine and chlorine in the diamond CVD process. Diam. Relat. Mater. 1999, 8, 231–235. [Google Scholar] [CrossRef]
- Vlassiouk, I.; Regmi, M.; Fulvio, P.; Dai, S.; Datskos, P.; Eres, G.; Smirnov, S. Role of Hydrogen in Chemical Vapor Deposition Growth of Large Single-Crystal Graphene. ACS Nano 2011, 5, 6069–6076. [Google Scholar] [CrossRef]
- Eddaoudi, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density By S. Lowell (Quantachrome Instruments, Boynton Beach), J.E. Shields (C. W. Post Campus of Long Island University), M.A. Thomas, and M. Thommes (Quantachrome In-struments). Kluwer Academic Publishers: Dordrecht, The Netherlands. 2004. xiv + 348 pp. $159.00. ISBN 1-4020-2302-2. J. Am. Chem. Soc. 2005, 127, 14117. [Google Scholar] [CrossRef]
- Haul, R.S.J.; Gregg, K.S.W. Sing: Adsorption, Surface Area and Porosity. 2. Auflage, Academic Press, London 1982. 303 Seiten, Preis: $ 49.50. Ber. Bunsenges. Phys. Chem. 1982, 86, 957. [Google Scholar] [CrossRef]
- Cui, X.; Bustin, R.; Dipple, G. Selective transport of CO2, CH4, and N2 in coals: Insights from modeling of experimental gas adsorption data. Fuel 2004, 83, 293–303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2 g−1) (a) | VT (cm3 g−1) (a) | Vmic (cm3 g−1) (b) | Vmeso (cm3 g−1) (c) | Average Pore Size (nm) (a) | CO2 Adsorption at 273 K (mmol g−1) | CO2 Adsorption at 298 K (mmol g−1) |
|---|---|---|---|---|---|---|---|
| HCFs | 211 | 0.406 | 0.027 | 0.379 | 7.693 | 0.818 | 0.485 |
| HCF-K | 279 | 0.182 | 0.128 | 0.054 | 2.619 | 0.496 | 0.311 |
| HCF-E | 379 | 0.322 | 0.171 | 0.151 | 3.398 | 0.810 | 0.385 |
| SPC | 158 | 0.146 | 0.073 | 0.073 | 3.680 | 0.204 | 0.208 |
| ST | 122 | 0.114 | 0.048 | 0.066 | 3.739 | 0.204 | 0.168 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero Valenzuela, A.E.; Chokradjaroen, C.; Thiangtham, S.; Saito, N. Hierarchical Porous Carbon Fibers Synthesized by Solution-Plasma-Generated Soot Deposition and Their CO2 Adsorption Capacity. Coatings 2022, 12, 1620. https://doi.org/10.3390/coatings12111620
Romero Valenzuela AE, Chokradjaroen C, Thiangtham S, Saito N. Hierarchical Porous Carbon Fibers Synthesized by Solution-Plasma-Generated Soot Deposition and Their CO2 Adsorption Capacity. Coatings. 2022; 12(11):1620. https://doi.org/10.3390/coatings12111620
Chicago/Turabian StyleRomero Valenzuela, Andres Eduardo, Chayanaphat Chokradjaroen, Satita Thiangtham, and Nagahiro Saito. 2022. "Hierarchical Porous Carbon Fibers Synthesized by Solution-Plasma-Generated Soot Deposition and Their CO2 Adsorption Capacity" Coatings 12, no. 11: 1620. https://doi.org/10.3390/coatings12111620