
Application of Vegetable Oil-Based Monomers in the Synthesis of Acrylic Latexes via Emulsion Polymerization

 , , , and
, , , and
Abstract
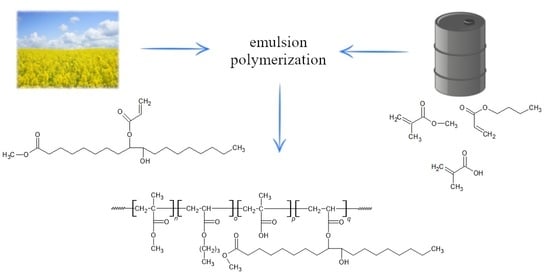
:
1. Introduction
2. Materials and Methods
2.1. Materials
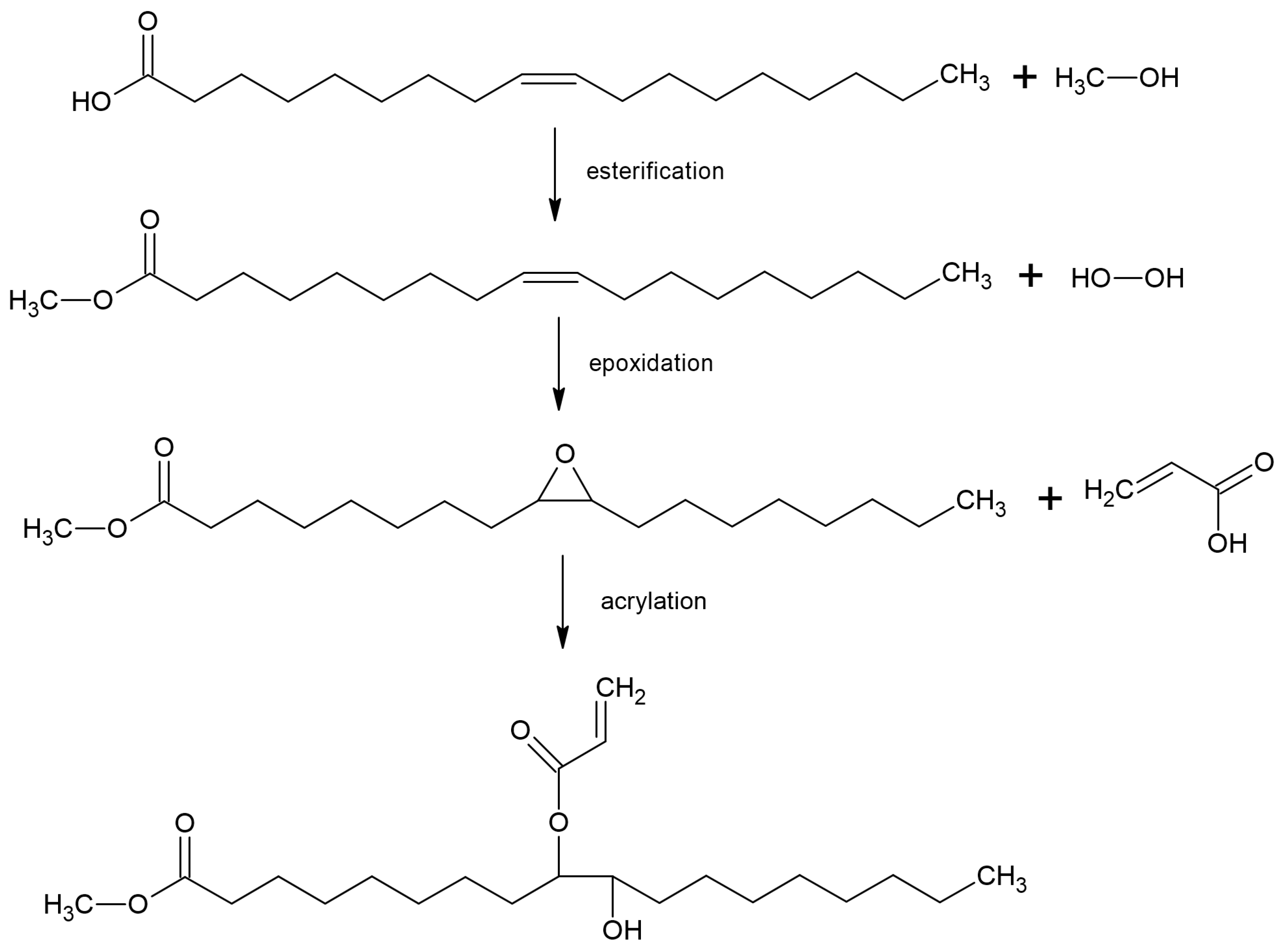
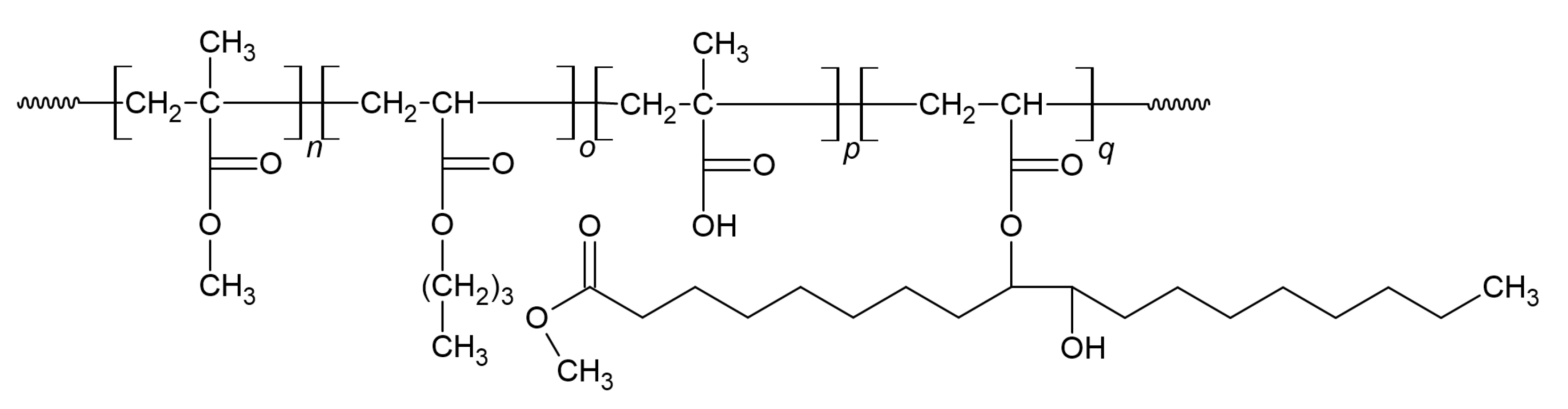
2.2. Synthesis and Characterization of Bio-Based Monomers
2.3. Synthesis and Characterization of Latexes
2.4. Preparation and Characterization of Coatings
3. Results and Discussion
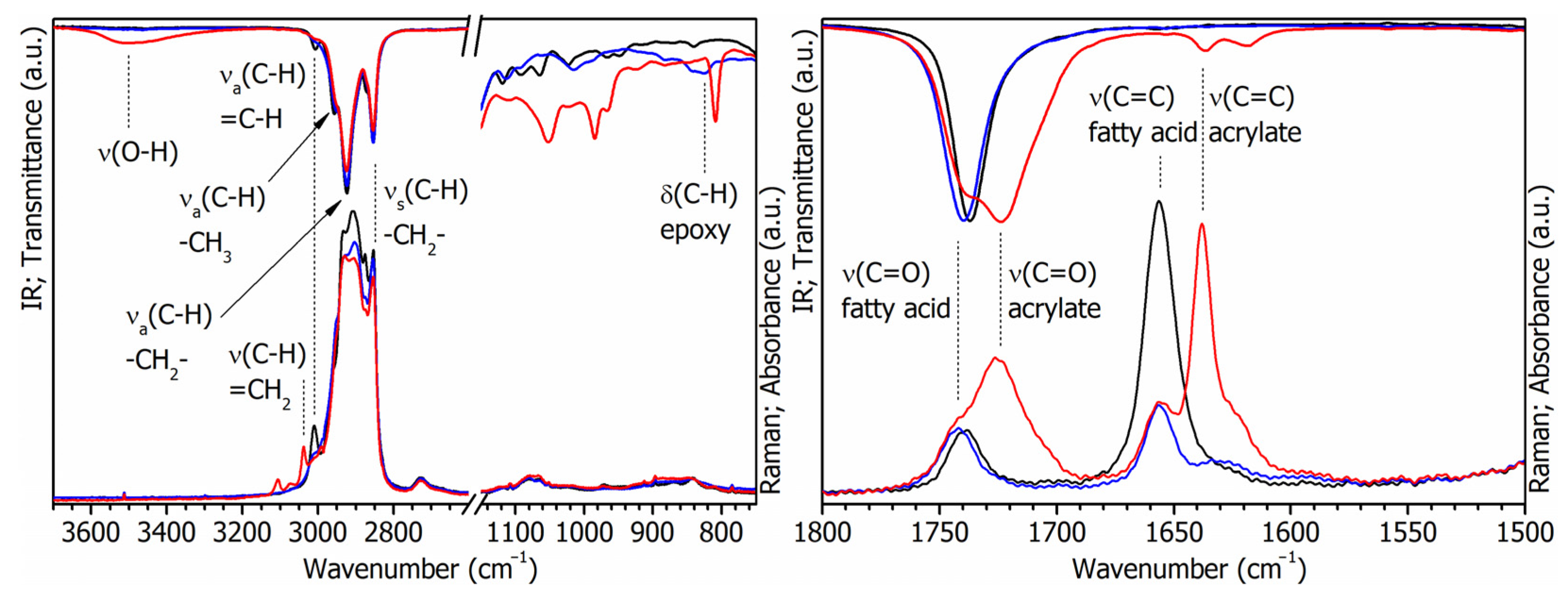
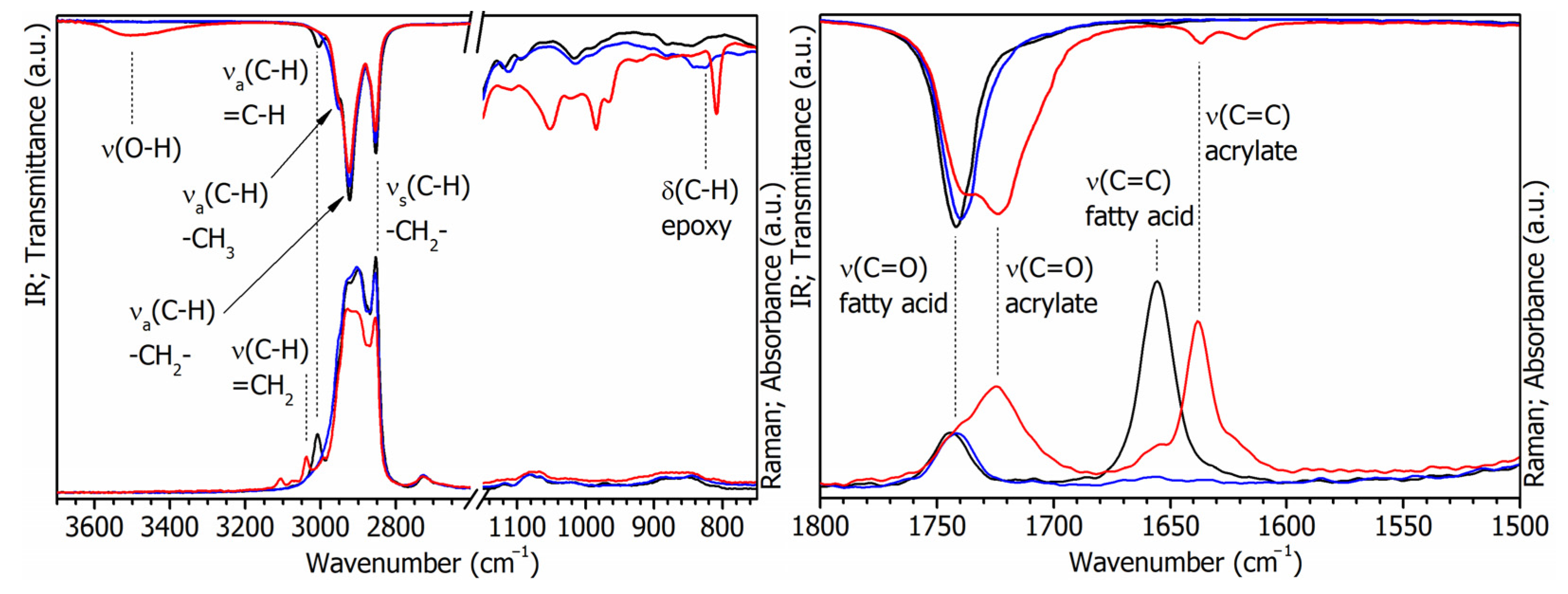
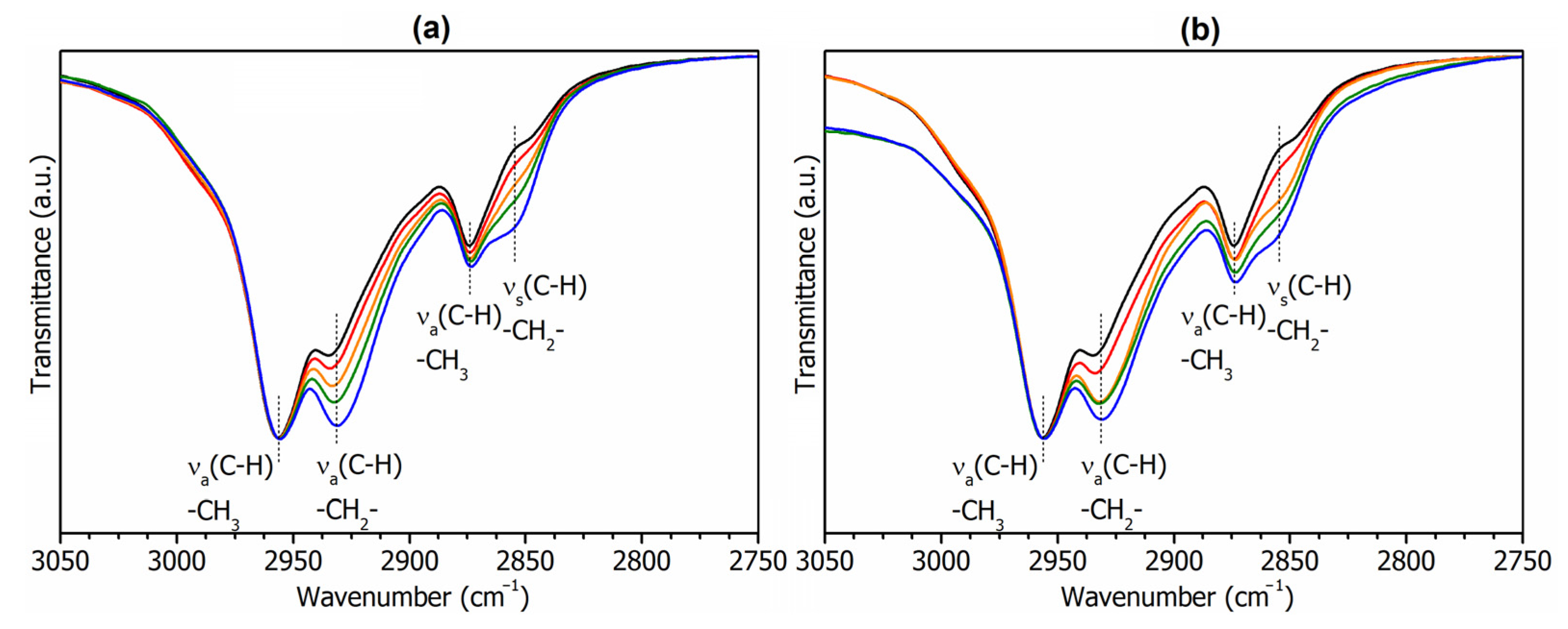
3.1. Characterization of Bio-Based Monomers
3.2. Characterization of Latexes
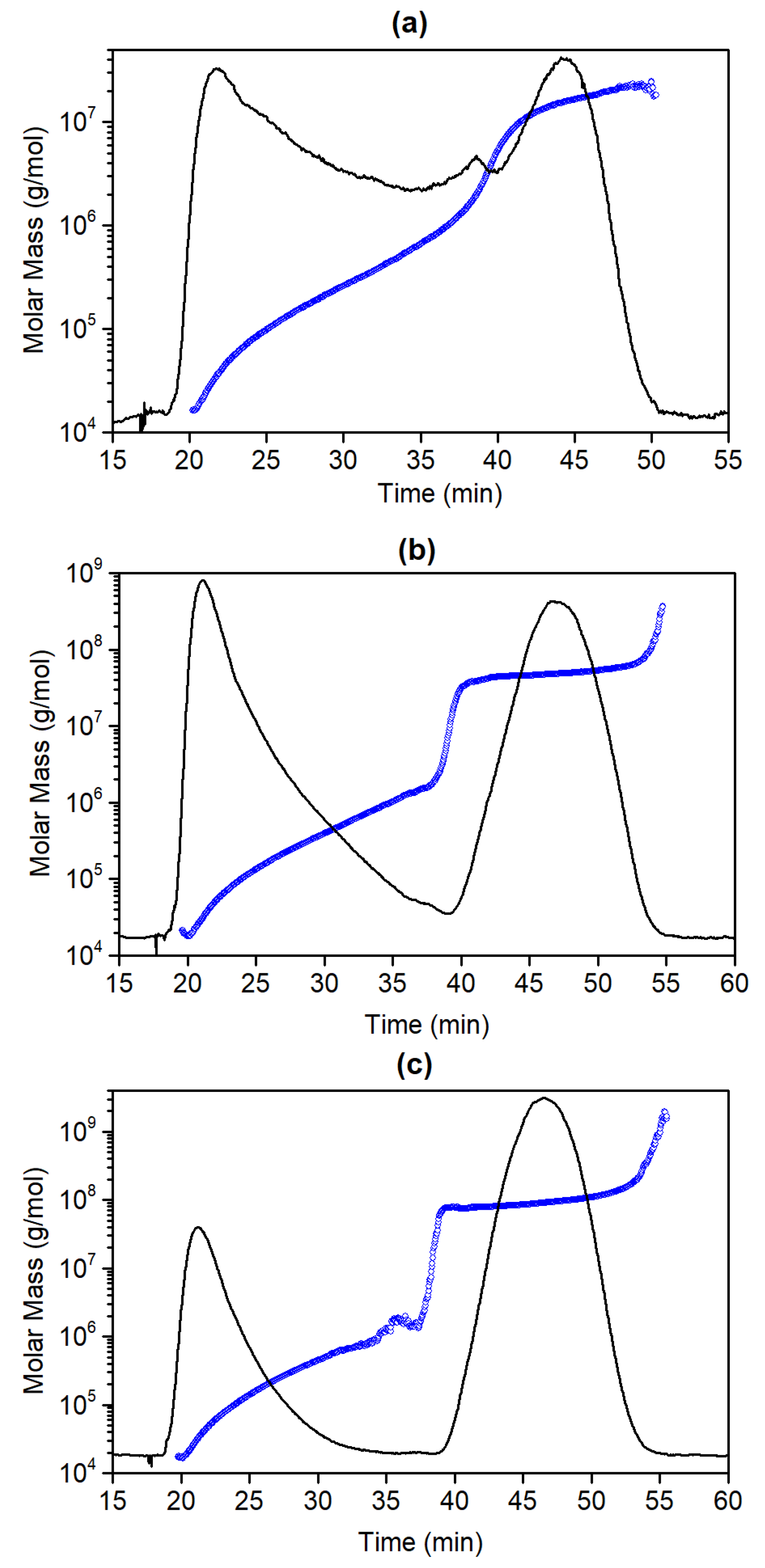
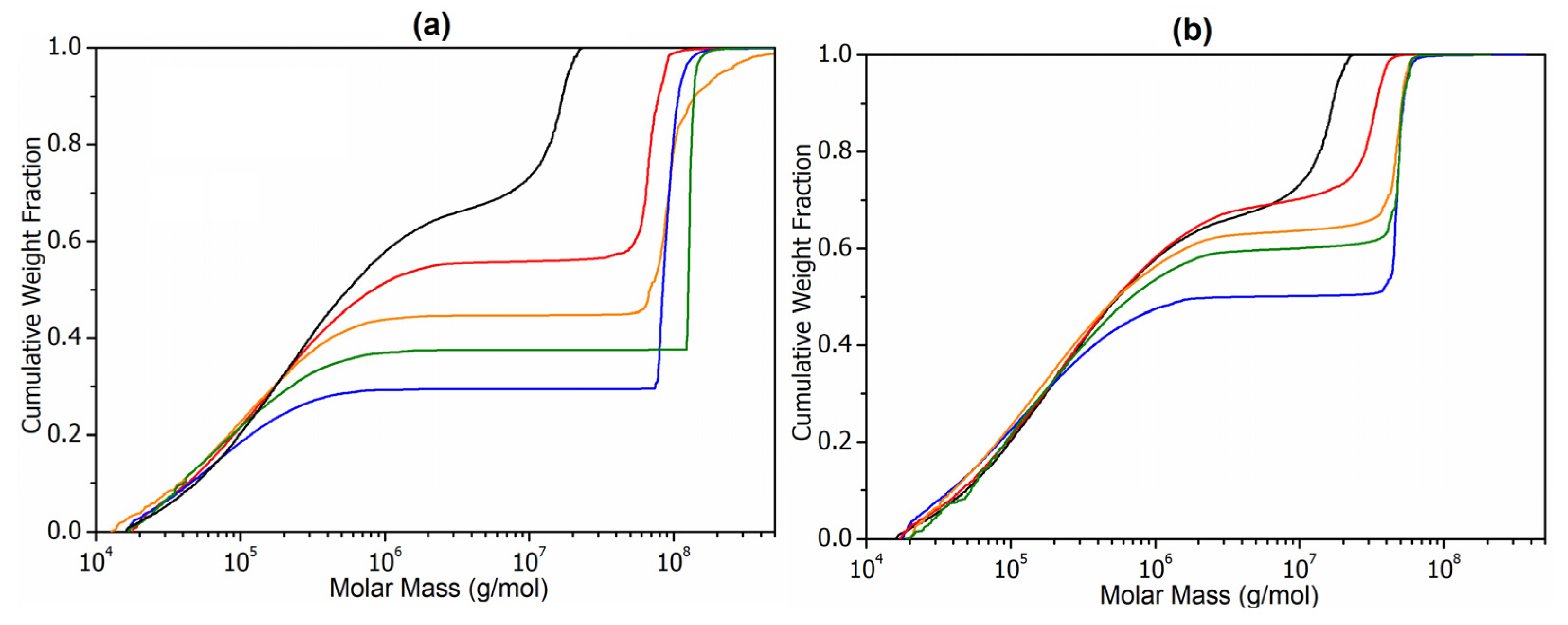
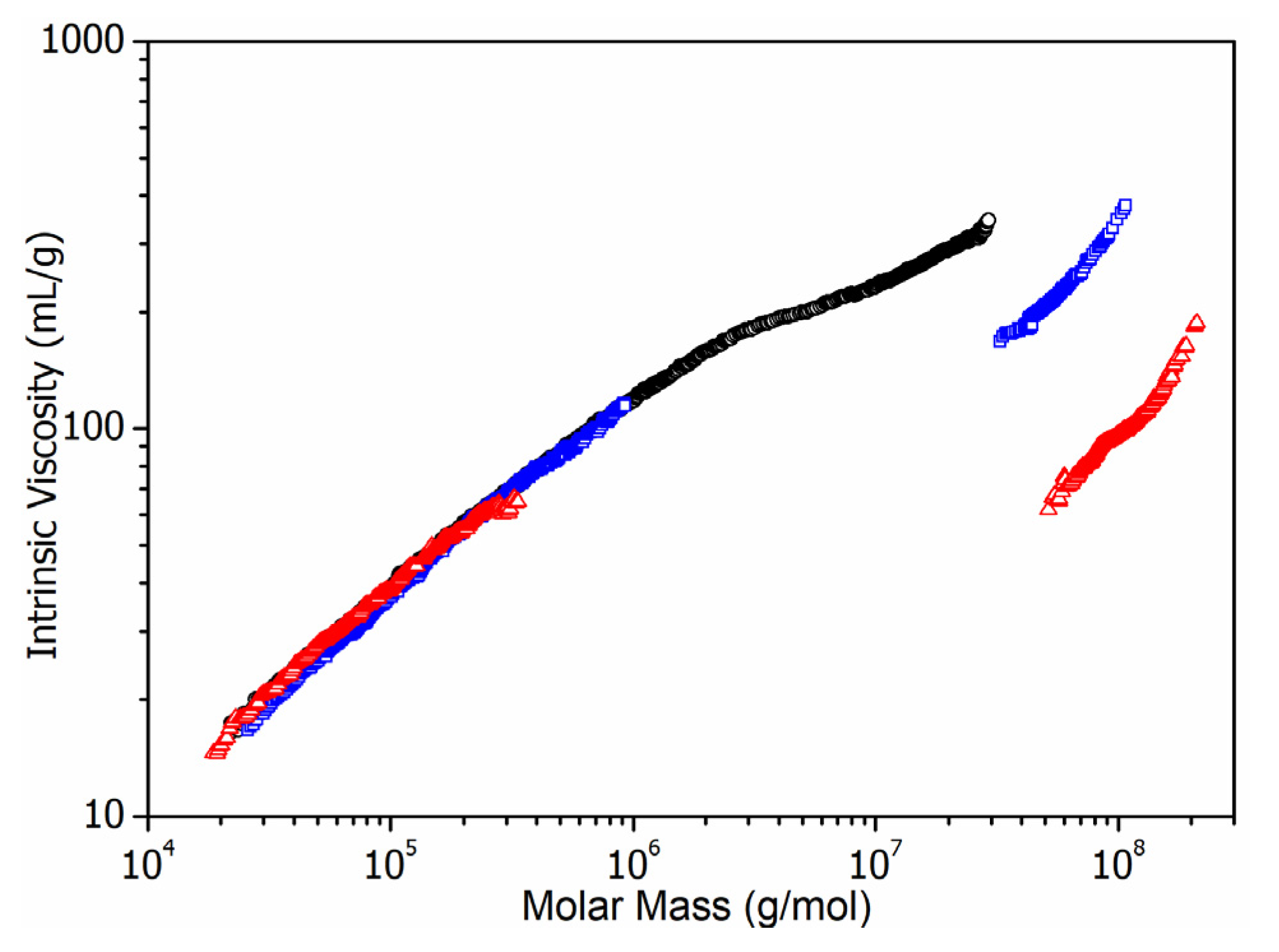
3.3. Molar Mass Determination of Latex Copolymers
3.4. Characterization of Coatings
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, C.S. Emulsion polymerization mechanisms and kinetics. Prog. Polym. Sci. 2006, 31, 443–486. [Google Scholar]
- Saldívar-Guerra, E.; Vivaldo-Lima, E. Introduction to polymers and polymer types. In Handbook of Polymer Synthesis, Characterization, and Processing; Saldívar-Guerra, E., Vivaldo-Lima, E., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; p. 8. ISBN 9780470630327. [Google Scholar]
- Iman, M.T.; Maji, K. Effect of crosslinker and nanoclay on starch and jute fabric-based green nanocomposites. Carbohydr. Polym. 2012, 89, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, K.G.; Arizaga, G.G.C.; Wypych, F. Biodegradable composites based on lignocellulosic fibers—An overview. Prog. Polym. Sci. 2009, 34, 982–1021. [Google Scholar] [CrossRef]
- Biswas, A.; Roy, M. Green products: An exploratory study on the consumer behaviour in emerging economies of the East. J. Cleaner Prod. 2015, 87, 463–468. [Google Scholar] [CrossRef]
- Lathi, P.S.; Mattiasson, B. Green approach for the preparation of biodegradable lubricant base stock from epoxidized vegetable oil. Appl. Catal. B 2007, 69, 207–212. [Google Scholar] [CrossRef]
- Montero de Espinosa, L.; Ronda, J.C.; Galià, M.; Cádiz, V. A new route to acrylate oils: Crosslinking and properties of acrylate triglycerides from high oleic sunflower oil. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 1159–1167. [Google Scholar] [CrossRef]
- Mandal, M.; Maji, T.K. Comparative study on the properties of wood polymer composites based on different modified soybean oils. J. Wood Chem. Technol. 2016, 37, 1–12. [Google Scholar] [CrossRef]
- Derksen, J.T.P.; Cuperus, F.P.; Kolster, P. Paints and coatings from renewable resources. Ind. Crops Prod. 1995, 3, 225–236. [Google Scholar] [CrossRef]
- Wuzella, G.; Mahendran, A.R.; Müller, U.; Kandelbauer, A. Photocrosslinking of an Acrylated Epoxidized Linseed Oil: Kinetics and its Application for Optimized Wood Coatings. J. Polym. Environ. 2012, 20, 1063–1074. [Google Scholar] [CrossRef]
- Montero de Espinosa, L.; Meier, M.A.R. Plant oils: The perfect renewable resource for polymer science?! Eur. Polym. J. 2001, 47, 837–852. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Erhan, S.Z. Ring-Opening Polymerization of Epoxidized Soybean Oil. J. Am. Oil Chem. Soc. 2010, 87, 437–444. [Google Scholar] [CrossRef]
- Wu, Y.; Li, K. Acrylated epoxidized soybean oil as a styrene replacement in a dicyclopentadiene-modified unsaturated polyester resin. J. Appl. Polym. Sci. 2018, 135, 46212. [Google Scholar] [CrossRef]
- Nekhavhambe, E.; Mukaya, H.E.; Bavon, D. Development of castor oil–based polymers: A review. J. Adv. Manuf. Process. 2019, 1, e10030. [Google Scholar] [CrossRef]
- Ho, Y.H.; Parthiban, A.; Thian, M.C.; Ban, Z.H.; Siwayanan, P. Acrylated Biopolymers Derived via Epoxidation and Subsequent Acrylation of Vegetable Oils. Int. J. Polym. Sci. 2022, 2022, 6210128. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, X.; Zhang, S.W. Biodegradable medical polymers: Fundamental Sciences. In Science and Principles of Biodegradable and Bioresorbable Medical Polymers—Materials and Properties, 1st ed.; Zhang, X., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 14–16. ISBN 978-0-08-100372-5. [Google Scholar]
- Netravali, A.N.; Pastore, C.M. Sustainable Composites—Fibers, Resins and Applications; DEStech Publication: Lancaster, PA, USA, 2015; p. 43. ISBN 978-1-60595-111-9. [Google Scholar]
- Teramoto, N. Synthetic Green Polymers from Renewable Monomers. In Handbook of Applied Biopolymer Technology—Synthesis, Degradation and Applications. Sharma, S.K., Mudhoo, A., Eds.; Royal Society of Chemistry (RSC): London, UK, 2011; pp. 24–29. ISBN 978-1-84973-151-5. [Google Scholar]
- Woźniak, E.; Waszkowska, E.; Zimny, T.; Sowa, S.; Twardowski, T. The rapeseed potential in Poland and Germany in the context of production, legislation, and intellectual property rights. Front. Plant Sci. 2019, 10, 1423. [Google Scholar] [CrossRef] [Green Version]
- Güner, F.S.; Yağci, Y.; Erciyes, A.T. Polymers from triglyceride oils. Prog. Polym. Sci. 2006, 31, 633–670. [Google Scholar] [CrossRef]
- Przybylski, R. Flax Oils and High Linolenic Oils. In Bailey's Industrial Oil and Fat Products, 6th ed.; Shahidi, F., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005; pp. 287–289. ISBN 9780471384601. [Google Scholar]
- Del Rio, E.; Galià, M.; Cádiz, V.; Lligadas, G.; Ronda, J.C. Polymerization of Epoxidized Vegetable Oil Derivatives: Ionic-Coordinative Polymerization of Methylepoxyoleate. J. Polym. Sci. A Polym. Chem. 2010, 48, 4995–5008. [Google Scholar] [CrossRef]
- Moreno, M.; Lampard, C.; Williams, N.; Lago, E.; Emmett, S.; Goikoetxea, M.; Barandiaran, M.J. Eco-paints from bio-based fatty acid derivative latexes. Prog. Org. Coat. 2015, 81, 101–106. [Google Scholar] [CrossRef]
- Crivello, J.V.; Narayan, R. Epoxidized Triglycerides as Renewable Monomers in Photoinitiated Cationic Polymerization. Chem. Mater. 1992, 4, 692–699. [Google Scholar] [CrossRef]
- Gandini, A.; Lacedra, T.M. From monomers to polymers from renewable resources: Recent advances. Prog. Polym. Sci. 2015, 48, 18–20. [Google Scholar] [CrossRef]
- Zhang, C.; Garrison, T.F.; Madbouly, S.A.; Kessler, M.R. Recent advances in vegetable oil-based polymers and their composites. Prog. Polym. Sci. 2017, 71, 91–143. [Google Scholar] [CrossRef]
- Neves, J.S.; Valadares, L.F.; Machado, F. Tailoring Acrylated Soybean Oil-Containing Terpolymers through Emulsion Polymerization. Colloids Interfaces 2018, 2, 46. [Google Scholar] [CrossRef] [Green Version]
- Knot, S.N.; Lascala, J.J.; Can, E.; Morye, S.S.; Williams, G.I.; Palmese, G.R.; Küsefoğlu, S.H.; Wool, R.P. Development and application of triglyceride-based polymers and composites. J. Appl. Polym. Sci. 2001, 82, 703–706. [Google Scholar]
- Scala, J.L.; Wool, R.P. Rheology of Chemically Modified Triglycerides. J. Appl. Polym. Sci. 2005, 95, 774–783. [Google Scholar] [CrossRef]
- Eren, T.; Küsefoğlu, S.H. Synthesis and Polymerization of the Bromoacrylated Plant Oil Triglycerides to Rigid, Flame-Retardant Polymers. J. Appl. Polym. Sci. 2004, 91, 2700–2710. [Google Scholar] [CrossRef]
- Wool, R.P.; Khot, S.N. Bio-Based Resins and Natural Fibers. In ASM Handbook, Volume 21—Composites; Miracle, D.B., Donaldson, S.L., Eds.; ASM International: Materials Park, OH, USA, 2001; pp. 185–186. ISBN 978-0-87170-703-1. [Google Scholar]
- Eren, T.; Küsefoğlu, S.H. Hydroxymethylation and polymerization of plant oil triglycerides. J. Appl. Polym. Sci. 2004, 91, 4037–4046. [Google Scholar] [CrossRef]
- Kollbe Ahn, B.; Kraft, S.; Wang, D.; Sun, X.S. Thermally Stable, Transparent, Pressure-Sensitive Adhesives from Epoxidized and Dihydroxyl Soybean Oil. Biomacromolecules 2011, 12, 1839–1843. [Google Scholar]
- Husić, S.; Javni, I.; Petrović, Z.S. Thermal and mechanical properties of glass reinforced soy-based polyurethane composites. Compos. Sci. Technol. 2005, 65, 19–25. [Google Scholar] [CrossRef]
- Johansson, K.; Johansson, M. A model study on fatty acid methyl esters as reactive diluents in thermally cured coil coating systems. Prog. Org. Coat. 2006, 55, 382–387. [Google Scholar] [CrossRef]
- Nameer, S.; Deltin, T.; Sundell, P.; Johansson, M. Bio-based multifunctional fatty acid methyl esters as reactive diluents in coil coatings. Prog. Org. Coat. 2019, 136, 105277. [Google Scholar] [CrossRef]
- Laurentino, L.S.; Medeiros, A.M.M.S.; Machado, F.; Costas, C.; Araújo, P.H.H.; Sayer, C. Synthesis of a biobased monomer derived from castor oil and copolymerization in an aqueous medium. Chem. Eng. Res. Des. 2018, 137, 213–220. [Google Scholar] [CrossRef]
- Lu, Y.; Larock, R.C. New hybrid latexes from a soybean oil-based waterborne polyurethane and acrylics via emulsion polymerization. Biomacromolecules 2007, 8, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Kohut, A.; Voronov, S.; Demchuk, Z.; Kirianchuk, V.; Kingsley, K.; Shevchuk, O.; Caillol, S.; Voronov, A. Non-Conventional Features of Plant Oil-Based Acrylic Monomers in Emulsion Polymerization. Molecules 2020, 25, 2990. [Google Scholar] [CrossRef]
- Kingsley, K.; Shevchuk, O.; Demchuk, Z.; Voronov, S.; Voronov, A. The features of emulsion copolymerization for plant oil-based vinyl monomers and styrene. Ind. Crops Prod. 2017, 109, 274–280. [Google Scholar] [CrossRef]
- Demchuk, Z.; Shevchuk, O.; Tarnavchyk, I.; Kirianchuk, V.; Lorenson, M.; Kohut, A.; Voronov, S.; Voronov, A. Free-Radical Copolymerization Behavior of Plant-Oil-Based Vinyl Monomers and Their Feasibility in Latex Synthesis. ACS Omega 2016, 1, 1374–1382. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, G.R.; Braquehais, J.R.; Da Silva, W.N.; Machado, F. Synthesis of soybean oil-based polymer lattices via emulsion polymerization process. Ind. Crops Prod. 2015, 65, 14–20. [Google Scholar] [CrossRef]
- Ladmiral, V.; Jeannin, R.; Lizarazu, K.P.; Lai-Kee-Him, J.; Bron, P.; Lacroix-Desmazes, P.; Caillol, S. Aromatic biobased polymer latex from cardanol. Eur. Polym. J. 2017, 93, 785–794. [Google Scholar] [CrossRef]
- Hanuš, J.; Unters, Z. Nahr. Genussm. Gebrauchsgegen-Staende 1901, 4, 913–920. [Google Scholar]
- Ling, H.; Junyan, L. Synthesis, modification and characterization of core–shell fluoroacrylate copolymer latexes. J. Fluorine Chem 2008, 129, 590–597. [Google Scholar] [CrossRef]
- Bunker, S.P.; Wool, R.P. Synthesis and Characterization of Monomers and Polymers for Adhesives from Methyl Oleate. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 451–458. [Google Scholar] [CrossRef]
- Jensen, A.T.; Sayer, C.; Araújo, P.H.; Machado, F. Emulsion copolymerization of styrene and acrylated methyl oleate. Eur. J. Lipid Sci. Technol. 2014, 116, 37–43. [Google Scholar] [CrossRef]
- Vávra, A.; Hájek, M.; Skopal, F. Acceleration and simplification of separation by addition of inorganic acid in biodiesel production. J. Cleaner Prod. 2018, 192, 390–395. [Google Scholar] [CrossRef]
- Hájek, M.; Vávra, A.; de Paz Carmona, H.; Kocík, J. The catalysed transformation of vegetable oils or animal fats to biofuels and bio-lubricants: A review. Catalysts 2021, 11, 1118. [Google Scholar] [CrossRef]
- Ugelstad, J.; El-Aasser, M.S.; Vanderhoff, J.W. Emulsion polymerization: Initiation of polymerization in monomer droplets. J. Polym. Sci. Polym. Lett. Ed. 1973, 11, 503–513. [Google Scholar] [CrossRef]
- Riess, G.; Labbe, C. Block copolymers in emulsion and dispersion polymerization. Macromol. Rapid Commun. 2004, 25, 401–435. [Google Scholar] [CrossRef]
- Kim, D.; Lee, D.Y.; Lee, K.; Choe, S. Effect of crosslinking agents on the morphology of polymer particles produced by one-step seeded polymerization. Macromol. Res. 2009, 17, 250–258. [Google Scholar] [CrossRef]
- Hergeth, W.D.; Lebek, W.; Stettin, E.; Witkowski, K.; Schmutzler, K. Particle formation in emulsion polymerization, 2. Aggregation of primary particles. Makromol. Chem. 1992, 193, 1607–1621. [Google Scholar] [CrossRef]
- Yilmaz, O.; Cheaburu, C.N.; Durraccio, D.; Gulumser, G.; Vasile, C. Preparation of stable acrylate/montmorillonite nanocomposite latex via in situ batch emulsion polymerization: Effect of clay types. Appl. Clay Sci. 2010, 49, 288–297. [Google Scholar] [CrossRef]
- Makan, A.C.; Williams, R.P.; Pasch, H. Field Flow Fractionation for the Size, Molar Mass, and Gel Content Analysis of Emulsion Polymers for Water-Based Coatings. Macromol. Chem. Phys. 2016, 217, 2027–2040. [Google Scholar] [CrossRef]
- Podzimek, S.; Machotova, J.; Snuparek, J.; Vecera, M.; Prokupek, L. Characterization of Molecular Structure of Acrylic Copolymers Prepared via Emulsion Polymerization Using A4F-MALS Technique. J. Appl. Polym. Sci. 2014, 131, 40995. [Google Scholar] [CrossRef]
- Podzimek, S. Asymmetric Flow Field Flow Fractionation. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9780471976707. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Bio-Based Monomer Content (wt.%) | Monomer (g) | ||||
|---|---|---|---|---|---|---|
| MMA | BA | MAA | AME_RO | AME_OA | ||
| REF | 0 | 21.00 | 28.00 | 1 | - | - |
| RO_5 | 5 | 19.93 | 26.57 | 1 | 2.5 | - |
| RO_10 | 10 | 18.86 | 25.14 | 1 | 5.0 | - |
| RO_15 | 15 | 17.79 | 23.71 | 1 | 7.5 | - |
| RO_20 | 20 | 16.72 | 22.28 | 1 | 10.0 | - |
| OA_5 | 5 | 19.93 | 26.57 | 1 | - | 2.5 |
| OA_10 | 10 | 18.86 | 25.14 | 1 | - | 5.0 |
| OA_15 | 15 | 17.75 | 23.71 | 1 | - | 7.5 |
| OA_20 | 20 | 16.72 | 22.28 | 1 | - | 10.0 |
| Fatty Acid Type | Content of Methyl Ester of Higher Fatty Acid (wt.%) | |||||
|---|---|---|---|---|---|---|
| ME_RO | EME_RO | AME_RO | ME_OA | EME_OA | AME_OA | |
| C16:0 | 5.7 | 4.5 | 3.6 | 7.2 | 6.7 | 5.8 |
| C18:0 | 2.3 | 1.4 | 1.2 | 2.1 | 1.9 | 1.6 |
| C18:1 | 57.4 | 10.4 | 8.3 | 76.2 | 11.2 | 8.8 |
| C18:2 | 26.4 | 1.2 | 0.9 | 13.9 | 0.6 | 0.2 |
| C18:3 | 6.2 | 0.1 | 0.1 | -* | -* | -* |
| C20:1 | 0.9 | 0.1 | 0.1 | 0.4 | -* | -* |
| others | 1.2 | 0.6 | 0.4 | 0.1 | 0.1 | 0.1 |
| Σ | 100.0 | 18.3 | 14.5 | 100.0 | 20.5 | 16.6 |
| Sample | ME_RO | EME_RO | AME_RO | ME_OA | EME_OA | AME_OA |
|---|---|---|---|---|---|---|
| Iodine value (g I2/100 g) | 108.2 ± 2.5 | 17.7 ± 1.4 | 54.1 ± 1.3 | 85.1 ± 2.1 | 7.5 ± 0.8 | 49.4 ± 2.2 |
| Sample | Coagulum (%) | Conversion (%) | Particle Size (nm) | Zeta Potential (mV) | ||
|---|---|---|---|---|---|---|
| Before Storing | After Storing | Before Storing | After Storing | |||
| REF | 1.7 ± 1.1 | 94.3 ± 1.0 | 102.1 ± 0.9 | 104.0 ± 1.6 | −42.2 ± 0.2 | −38.5 ± 1.1 |
| RO_5 | 3.3 ± 1.4 | 95.0 ± 0.7 | 100.2 ± 1.3 | 102.3 ± 1.1 | −44.4 ± 0.9 | −41.5 ± 0.6 |
| RO_10 | 3.8 ± 0.4 | 93.1 ± 1.0 | 108.2 ± 1.3 | 111.4 ± 1.0 | −44.8 ± 0.3 | −42.3 ± 0.8 |
| RO_15 | 5.3 ± 0.4 | 93.9 ± 1.2 | 102.5 ± 1.0 | 106.8 ± 0.8 | −40.3 ± 0.6 | −41.7 ± 1.0 |
| RO_20 | 21.3 ± 4.6 | 95.4 ± 1.9 | 96.3 ± 0.4 | 101.5 ± 0.9 | −40.3 ± 0.8 | −39.6 ± 0.4 |
| OA_5 | 4.9 ± 1.0 | 95.6 ± 0.8 | 105.0 ± 1.6 | 106.9 ± 1.3 | −40.3 ± 1.7 | −41.2 ± 0.9 |
| OA_10 | 5.3 ± 1.2 | 94.7 ± 0.9 | 101.1 ± 1.7 | 105.1 ± 0.7 | −46.1 ± 0.5 | −40.2 ± 0.6 |
| OA_15 | 7.2 ± 2.5 | 96.7 ± 1.8 | 95.2 ± 1.2 | 99.9 ± 1.5 | −42.6 ± 1.0 | −41.0 ± 1.3 |
| OA_20 | 18.9 ± 6.9 | 95.5 ± 1.2 | 90.3 ± 1.0 | 96.7 ± 0.6 | −44.0 ± 1.0 | −39.0 ± 0.8 |
| Sample | ||||
|---|---|---|---|---|
| REF | 170 | 6.1 | 18 | 36 |
| RO_5 | 140 | 31.4 | 83 | 224 |
| RO_10 | 120 | 119.7 | 3360 | 998 |
| RO_15 | 160 | 81.4 | 133 | 509 |
| RO_20 | 170 | 67.0 | 112 | 394 |
| OA_5 | 150 | 9.6 | 31 | 64 |
| OA_10 | 140 | 17.3 | 48 | 124 |
| OA_15 | 150 | 19.7 | 49 | 131 |
| OA_20 | 130 | 24.5 | 50 | 188 |
| Sample | Gloss 60° (GU) | Pull Strength (MPa) | WCA (°) | Tg (°C) |
|---|---|---|---|---|
| REF | 83.5 ± 0.1 | 3.3 ± 0.3 | 64.9 ± 1.5 | 1.3 ± 0.3 |
| RO_5 | 83.8 ± 0.2 | 3.4 ± 0.2 | 66.6 ± 4.0 | −2.7 ± 0.4 |
| RO_10 | 83.7 ± 0.1 | 3.2 ± 0.2 | 72.6 ± 2.3 | −5.3 ± 0.4 |
| RO_15 | 84.2 ± 0.1 | 3.1 ± 0.3 | 70.6 ± 3.6 | −8.2 ± 0.3 |
| RO_20 | 83.7 ± 0.2 | 3.1 ± 0.2 | 66.7 ± 1.2 | −12.4 ± 1.0 |
| OA_5 | 83.5 ± 0.2 | 3.2 ± 0.3 | 67.7 ± 1.4 | −2.6 ± 0.2 |
| OA_10 | 83.1 ± 0.3 | 3.2 ± 0.4 | 70.2 ± 0.6 | −5.7 ± 0.6 |
| OA_15 | 83.4 ± 0.1 | 3.2 ± 0.2 | 73.3 ± 7.3 | −7.5 ± 0.3 |
| OA_20 | 84.1 ± 0.1 | 3.1 ± 0.2 | 76.2 ± 0.7 | −10.8 ± 0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolář, M.; Machotová, J.; Hájek, M.; Honzíček, J.; Hájek, T.; Podzimek, Š. Application of Vegetable Oil-Based Monomers in the Synthesis of Acrylic Latexes via Emulsion Polymerization. Coatings 2023, 13, 262. https://doi.org/10.3390/coatings13020262
Kolář M, Machotová J, Hájek M, Honzíček J, Hájek T, Podzimek Š. Application of Vegetable Oil-Based Monomers in the Synthesis of Acrylic Latexes via Emulsion Polymerization. Coatings. 2023; 13(2):262. https://doi.org/10.3390/coatings13020262
Chicago/Turabian StyleKolář, Martin, Jana Machotová, Martin Hájek, Jan Honzíček, Tomáš Hájek, and Štěpán Podzimek. 2023. "Application of Vegetable Oil-Based Monomers in the Synthesis of Acrylic Latexes via Emulsion Polymerization" Coatings 13, no. 2: 262. https://doi.org/10.3390/coatings13020262
APA StyleKolář, M., Machotová, J., Hájek, M., Honzíček, J., Hájek, T., & Podzimek, Š. (2023). Application of Vegetable Oil-Based Monomers in the Synthesis of Acrylic Latexes via Emulsion Polymerization. Coatings, 13(2), 262. https://doi.org/10.3390/coatings13020262