
Polymer Wrapping onto Nanoparticles Induces the Formation of Hybrid Colloids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. A Brief Historical Background
2. Introduction
3. The Model
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Obituary
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clutton-Brock, J. Domesticated Animals from Early Times; Heinemann: London, UK, 1981. [Google Scholar]
- Posthumus, L. Hybrid Monsters in the Classical World. The Nature and Function of Hybrid Monsters in Greek Mythology, Literature and Art. Ph.D. Thesis, University of Stellenbosch, Stellenbosch, South Africa, 2011. [Google Scholar]
- N.B. The horrible hybrid figures imagined by Hieronymus Bosch (1453–1516) and his followers, are built-up according to similar rules. Bosch’s seminal activity inspired artists and artisans, whose operas are collected in wunder-kammers.
- Linnaeus, C. Systema Naturae, Sive Regna tria Naturae Systematice Proposita per Classes, Ordines, Genera, & Species; Haak: Leiden, The Netherlands, 1735; pp. 1–12. [Google Scholar]
- Burke, J.M.; Arnold, M.M. Genetics and the fitness of hybrids. Annu. Rev. Genet. 2001, 35, 31–52. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. IUPAC. Compendium of Chemical Terminology, 2nd ed.; The “Gold Book”; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry; Oxford University Press: London, UK, 1939. [Google Scholar]
- de Gennes, P.G. Soft Matter (Nobel Lecture). Angew. Chem. Int. Ed. 1992, 31, 842–845. [Google Scholar] [CrossRef]
- Thunugunta, T.; Reddy, A.C.; Lakshmana, R.D.C. Green synthesis of nanoparticles: Current prospectus. Nanotechnol. Rev. 2015, 4, 303–323. [Google Scholar] [CrossRef]
- Win, K.Y.; Feng, S.-S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles fo oral delivery of anticancer drugs. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Murray, C.B.; Weller, D.; Folks, L.; Moser, A. Monodisperse FePt Nanoparticles and Ferromagnetic FePt Nanocrystal Superlattices. Science 2000, 287, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Mohammed-Saied, W.; Kaur, R.; Badea, I. Nanoparticles in gene therapy: From design to clinical applications. Rev. Nanosci. Nanotechnol. 2013, 2, 275–299. [Google Scholar] [CrossRef]
- La Mesa, C.; Risuleo, G. Stabilization of Food Colloids: The Role of Electrostatic and Steric Forces. In Some New Aspects of Colloidal Systems in Food; Milani, J.M., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Li, N.; Zeng, S.; He, L.; Zhong, W. Probing Nanoparticle-Protein Interaction by Capillary Electrophoresis. Anal. Chem. 2010, 82, 7460–7466. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, K.; Kang, E.-C.; Karumada, S.; Sunamoto, J.; Principi, T.; Winnik, F.M. Controlled Association of Amphphilic Polymers in Water; Thermosensitive Nanoparticles Formed by Self-Assembly of Hydrophobically Modified Pullulan and poly(N-isopropylacrylamides). Macromolecules 2000, 33, 3244–3249. [Google Scholar] [CrossRef]
- Jee, A.-Y.; Tsang, B.; Granick, S. Colloidal phase transitions. A switch for phase shifting. Nat. Mater. 2015, 14, 17–18. [Google Scholar] [CrossRef]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernandez, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface Functionalization of Nanoparticles with Polyethylene Glycol: Effects on Protein Adsorption and Cellular Uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef]
- Arslan, M.; Gevrek, T.N.; Lyskawa, J.; Szunerits, S.; Boukherroub, R.; Sanyal, R.; Woisel, P.; Sanyal, A. Bioinspired Anchorable Thiol-Reactive Polymers: Synthesis and Applications toward Surface Functionalization of Magnetic Nanoparticles. Macromolecules 2014, 47, 5124–5134. [Google Scholar] [CrossRef]
- Sehlleier, Y.H.; Abdali, A.; Huelser, T.; Wiggers, H.; Schulz, C. Functionalization of SiO2 nanoparticles and their super-hydrophobic surface coating. Spec. Publ. R. Soc. Chem. 2012, 336, 113–120. [Google Scholar]
- Demin, A.M.; Vigorov, A.Y.; Nizova, I.A.; Uimin, M.A.; Shchegoleva, N.N.; Ermakov, A.E.; Krasnov, V.P.; Charushin, V.N. Functionalization of Fe3O4 magnetic nanoparticles with RGD peptide derivatives. Mendeleev Commun. 2014, 24, 20–22. [Google Scholar] [CrossRef]
- Vauthier, C.; Labarre, D. Modular biomimetic drug delivery systems. J. Drug Deliv. Sci. Technol. 2008, 18, 59–68. [Google Scholar] [CrossRef]
- Frey, N.A.; Peng, S.; Cheng, K.; Sun, S. Magnetic nanoparticles: Synthesis, functionalization, and applications in bioimaging and magnetic energy storage. Chem. Soc. Rev. 2009, 38, 2532–2542. [Google Scholar] [CrossRef]
- Epple, M.; Ganesan, K.; Heumann, R.; Klesing, J.; Kovtun, A.; Neumann, S.; Sokolovs, V. Application of calcium phosphate nanoparticles in biomedicine. J. Mater. Chem. 2010, 20, 18–23. [Google Scholar] [CrossRef]
- Xing, X.; Hua, L.; Ngai, T. Depletion versus stabilization induced by polymers and nanoparticles: The state of the art. Curr. Opin. Colloid Interface Sci. 2015, 20, 54–59. [Google Scholar] [CrossRef]
- Ji, S.; Walz, J.Y. Depletion Flocculation Induced by Synergistic Effects of Nanoparticles and Polymers. J. Phys. Chem. B 2013, 117, 16602–16609. [Google Scholar] [CrossRef]
- Burger, S.; Bartsch, E. Influence of the polymer size on depletion attraction-induced gel and glass transitions of microgel colloids. Colloids Surf. A Physicochem. Eng. Asp. 2014, 442, 6–15. [Google Scholar] [CrossRef]
- Tardani, F.; La Mesa, C. Attempts to control depletion in the surfactant-assisted stabilization of single- walled carbon nanotubes. Colloids Surf. A Physicochem. Eng. Asp. 2014, 443, 123–128. [Google Scholar] [CrossRef]
- Fujigaya, T. Development of polymer-wrapping methods for functionalization of carbon materials. Polym. J. 2023, 55, 181–191. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Rubinstein, M. Adsorption of hydrophobic poly-electrolytes at oppositely charges surfaces. Macromolecules 2002, 35, 2754–2768. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Zhulina, E.B.; Rubinstein, M. Structure of adsorbed polyampholyte layers at charged objects. Macromolecules 2001, 34, 627–639. [Google Scholar] [CrossRef]
- Louguet, S.; Kumar, A.C.; Guidolin, N.; Sigaud, G.; Duguet, E.; Lecommandoux, S.; Shatz, C. Control of the PEO Chain Conformation on Nanoparticles by Adsorption of PEO-block-Poly(L-lysine) Copolymers and Its Sigificance on Colloidal Stability and Protein Repellency. Langmuir 2011, 27, 12891–12901. [Google Scholar] [CrossRef]
- Kusano, T.; Kumano, N.; Yoshimune, W.; Munekata, T.; Matsunaga, T.; Harada, M. Interplay between Interparticle Potential and Adsorption Structure in Nanoparticle Dispersions with Polymer Addition as Displayed by Small-Angle Scattering. Langmuir 2021, 37, 7503–7512. [Google Scholar] [CrossRef] [PubMed]
- Vold, R.D.; Vold, M.J. Colloid and Interface Chemistry; Addison-Wesley: Reading, MA, USA, 1983; Volume IV, pp. 134–148. [Google Scholar]
- Adamson, A.W. Physical Chemistry of Surfaces, Vth ed.; Wiley: New York, NY, USA, 1990; Volume IX, pp. 421–433. [Google Scholar]
- Suzuki, A.; Hara, T. Kinetics of one-dimensional swelling and shrinking of polymer gels under mechanical constraint. J. Chem. Phys. 2001, 114, 5012–5015. [Google Scholar] [CrossRef]
- Yagihara, S.; Hikichi, K. Cooperative Interaction on Side-Chain Motion of Poly(α-amino-acid). Polym. J. 1982, 14, 233–240. [Google Scholar] [CrossRef]
- de Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Malmsten, M.; Linse, P.; Cosgrove, T. Adsorption of PEO-PPO-PEO block copolymers at silica. Macromolecules 1992, 25, 2474–2481. [Google Scholar] [CrossRef]
- Nativ-Roth, E.; Shvartzman-Cohen, R.; Bounioux, C.; Florent, M.; Zhang, D.; Szleifer, I.; Yerushalmi-Rozen, R. Physical Adsorption of Block Copolymers to SWNT and MWNT: A Nonwrapping Mechanism. Macromolecules 2007, 40, 3676–3685. [Google Scholar] [CrossRef]
- Dorris, A.; Rucareanu, S.; Reven, L.; Barrett, C.J.; Lennox, R.B. Preparation and Characterization of Polyelectrolyte-coated Gold Nanoparticles. Langmuir 2008, 24, 2532–2538. [Google Scholar] [CrossRef]
- Bolukbasi, I. Tailoring Nanoparticles and Polymers for Cooperative Interfacial and Surface Interactions. Ph.D. Thesis, University of Massachusetts, Amherst, MA, USA, 2014. [Google Scholar]
- Schroffenegger, M.; Leitner, N.S.; Morgese, G.; Ramakrishna, S.N.; Willinger, M.; Benetti, E.M.; Reimhult, E. Polymer Topology Determines the Formation of Protein Corona on Core–Shell Nanoparticles. ACS Nano 2020, 14, 12708–12718. [Google Scholar] [CrossRef] [PubMed]
- Robson, R.J.; Dennis, E.A. The Size, Shape, and Hydration of Nonionic Surfactant Micelles. Triton X-100. J. Phys. Chem. 1977, 81, 1075–1078. [Google Scholar] [CrossRef]
- Long, J.A.; Rankin, B.M.; Ben-Amotz, D. Micelle Structure and Hydrophobic Hydration. J. Am. Chem. Soc. 2015, 137, 10809–10815. [Google Scholar]
- Quant, C.A.; Marla, K.T.; Meredith, J.C. Osmotic pressure and chemical potential of silica nanoparticles in aqueous poly(ethyleneoxide) solution. Colloids Surf. A Physicochem. Eng. Asp. 2008, 317, 129–135. [Google Scholar] [CrossRef]
- Eirich, F.R. The conformational states of macromolecules adsorbed at solid-liquid interfaces. J. Colloid Interface Sci. 1977, 58, 423–436. [Google Scholar] [CrossRef]
- Karlstroem, G. A new model for upper and lower critical solution temperatures in poly(ethylene oxide) solutions. J. Phys. Chem. 1985, 89, 4962–4964. [Google Scholar] [CrossRef]
- Docter, D.; Westmeier, D.; Markiewicz, M.; Stolte, S.; Knauer, S.K.; Stauber, R.H. The nanoparticle biomolecule corona: Lessons learned-challenge accepted? Chem. Soc. Rev. 2015, 44, 6094–6121. [Google Scholar] [CrossRef]
- Gref, R.; Luck, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnish, S.; Blunk, T.; Muller, R.H. Stealth corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Louguet, S.; Kumar, A.C.; Guidolin, N.; Sigaud, G.; Duguet, E.; Lecommandoux, S.; Schatz, C. A physico-chemical investigation of poly(ethylene oxide)-block poly(L-lysine) copolymer adsorption onto silica nanoparticles. J. Colloid Interface Sci. 2011, 359, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Sek, L.; Boyd, B.J.; Charman, W.N.; Porter, C.J. Examination of the impact of a range of Pluronic surfactants on the in-vivo solubilization behaviour and oral bioavailability of lipidic formulations of atovaquone. J. Pharm. Pharmacol. 2006, 58, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Lew, T.T.S.; Wong, M.H.; Kwak, S.-Y.; Sinclair, R.; Koman, V.B.; Strano, M.S. Rational Design Principles for the Transport and Subcellular Distribution of Nanomaterials into Plant Protoplasts. Small 2018, 14, 1802086. [Google Scholar] [CrossRef]
- Dai, J.; Dong, X.; Wang, Q.; Lou, X.; Xia, F.; Wang, S. PEG-Polymer Encapsulated Aggregation-Induced Emission Nanoparticles for Tumor Theranostics. Adv. Healthc. Mater. 2021, 10, 2101036. [Google Scholar] [CrossRef] [PubMed]
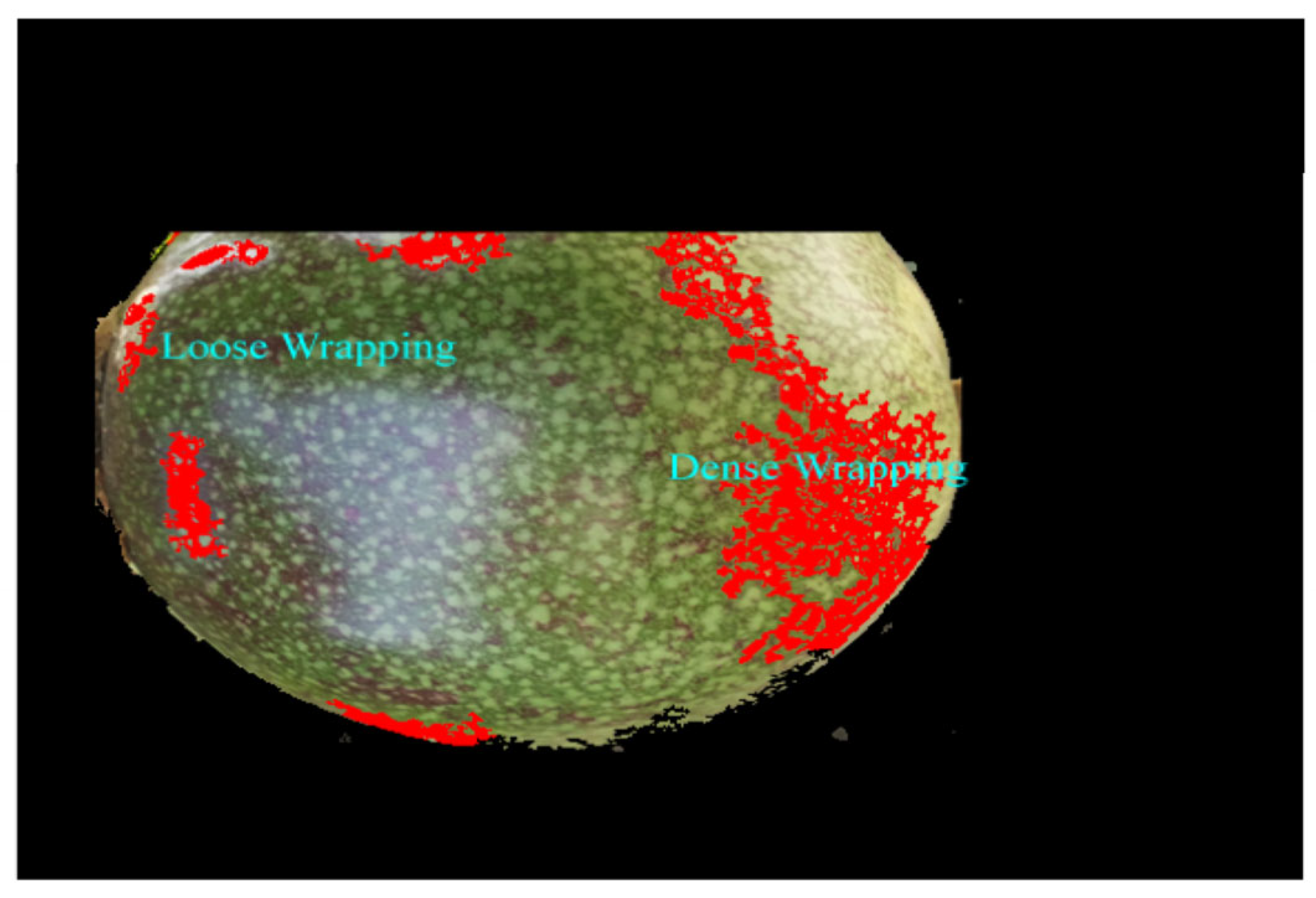

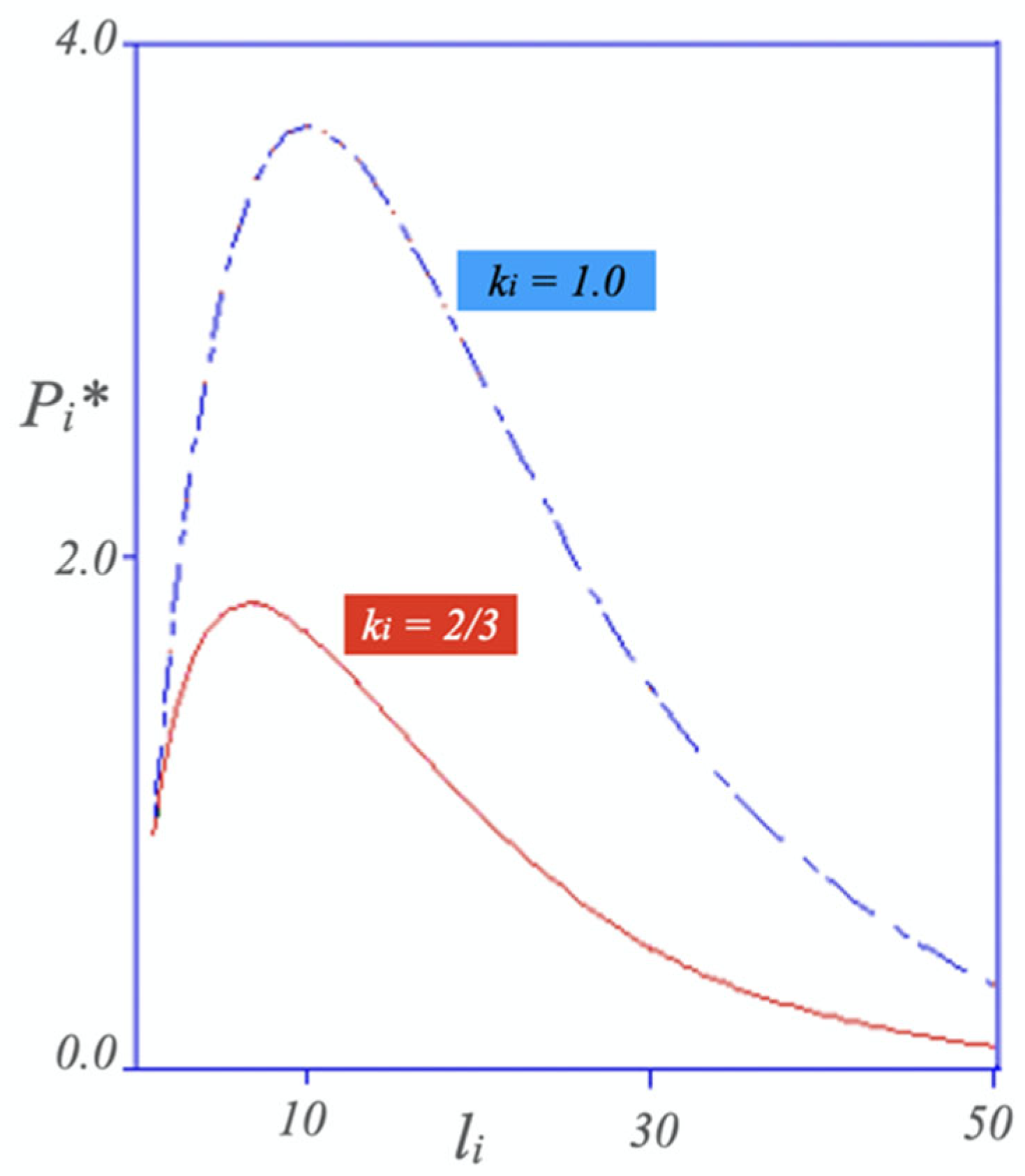
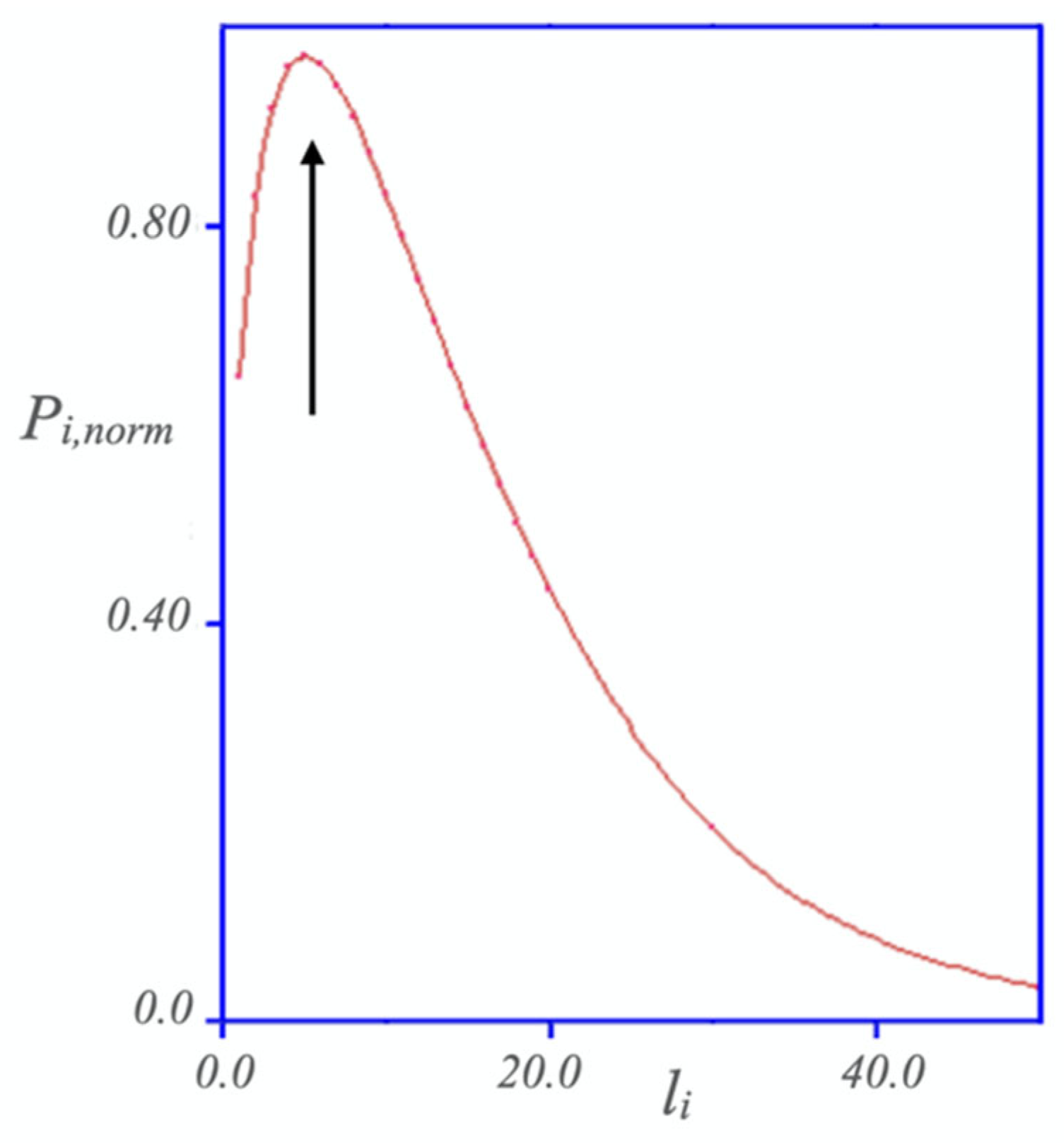
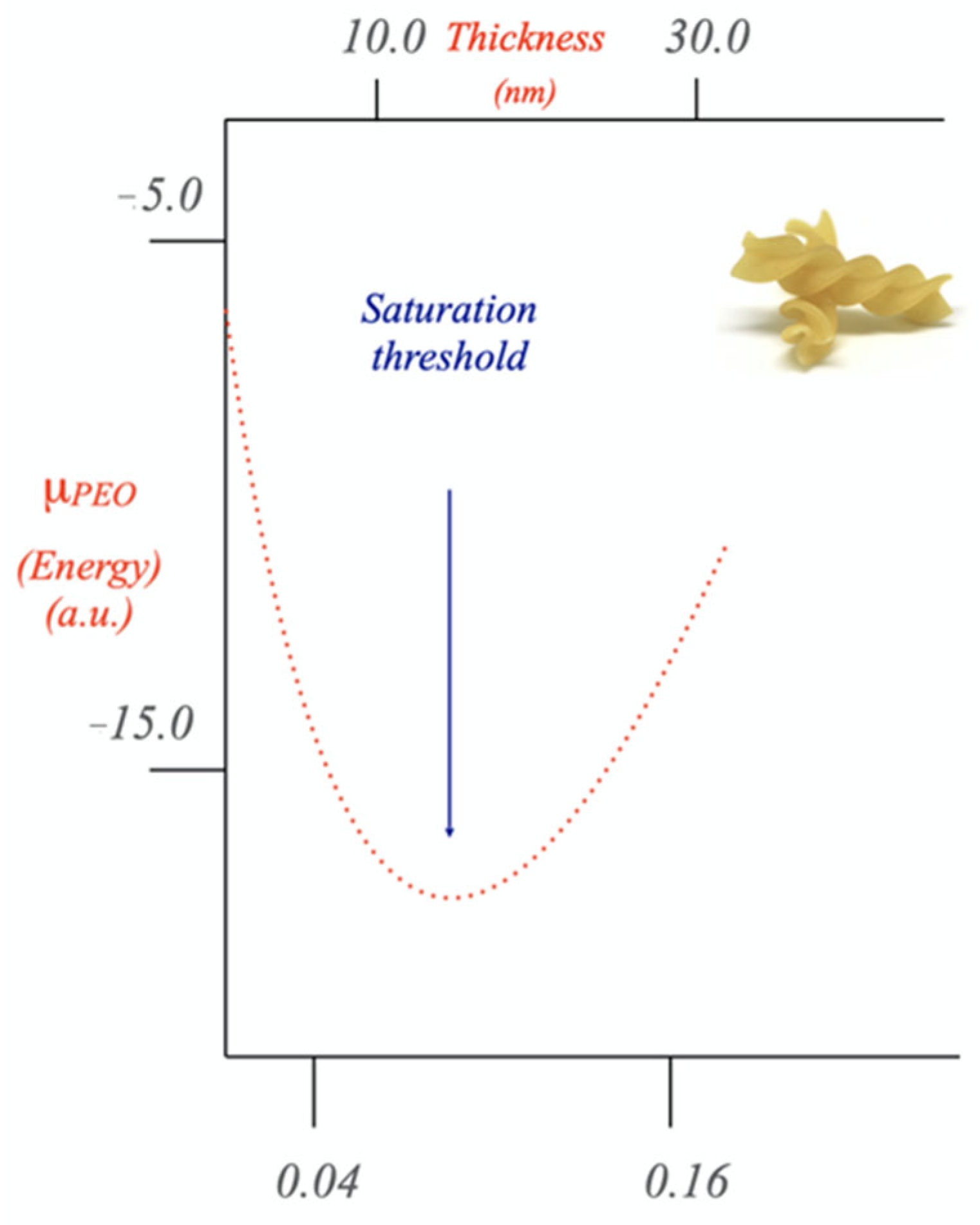
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Mesa, C.; Risuleo, G. Polymer Wrapping onto Nanoparticles Induces the Formation of Hybrid Colloids. Coatings 2023, 13, 823. https://doi.org/10.3390/coatings13050823
La Mesa C, Risuleo G. Polymer Wrapping onto Nanoparticles Induces the Formation of Hybrid Colloids. Coatings. 2023; 13(5):823. https://doi.org/10.3390/coatings13050823
Chicago/Turabian StyleLa Mesa, Camillo, and Gianfranco Risuleo. 2023. "Polymer Wrapping onto Nanoparticles Induces the Formation of Hybrid Colloids" Coatings 13, no. 5: 823. https://doi.org/10.3390/coatings13050823