
Polypyrrole/α-Fe2O3 Hybrids for Enhanced Electrochemical Sensing Performance towards Uric Acid

 , and
, and
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Solvents
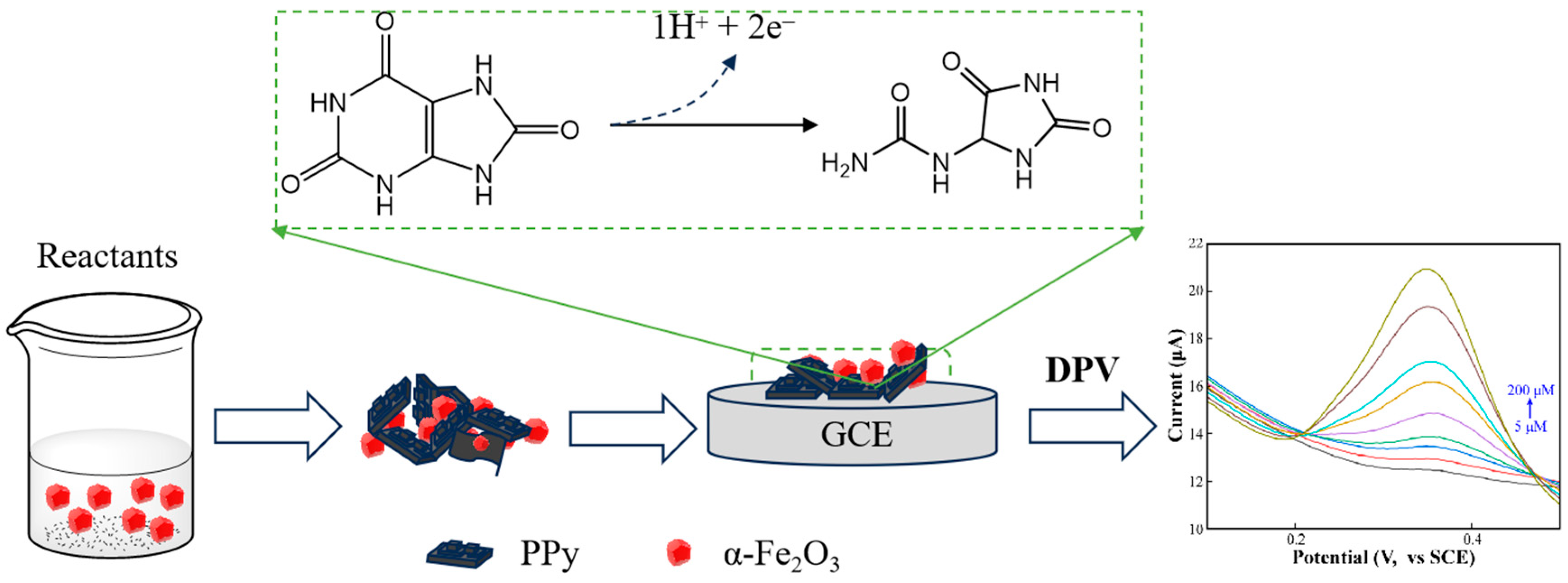
2.2. The Fabrication of PPy/α-Fe2O3 Composite
2.3. Characterization
2.4. Preparation of Working Electrodes
2.5. Biosensing Performance Measurements
3. Results and Discussion
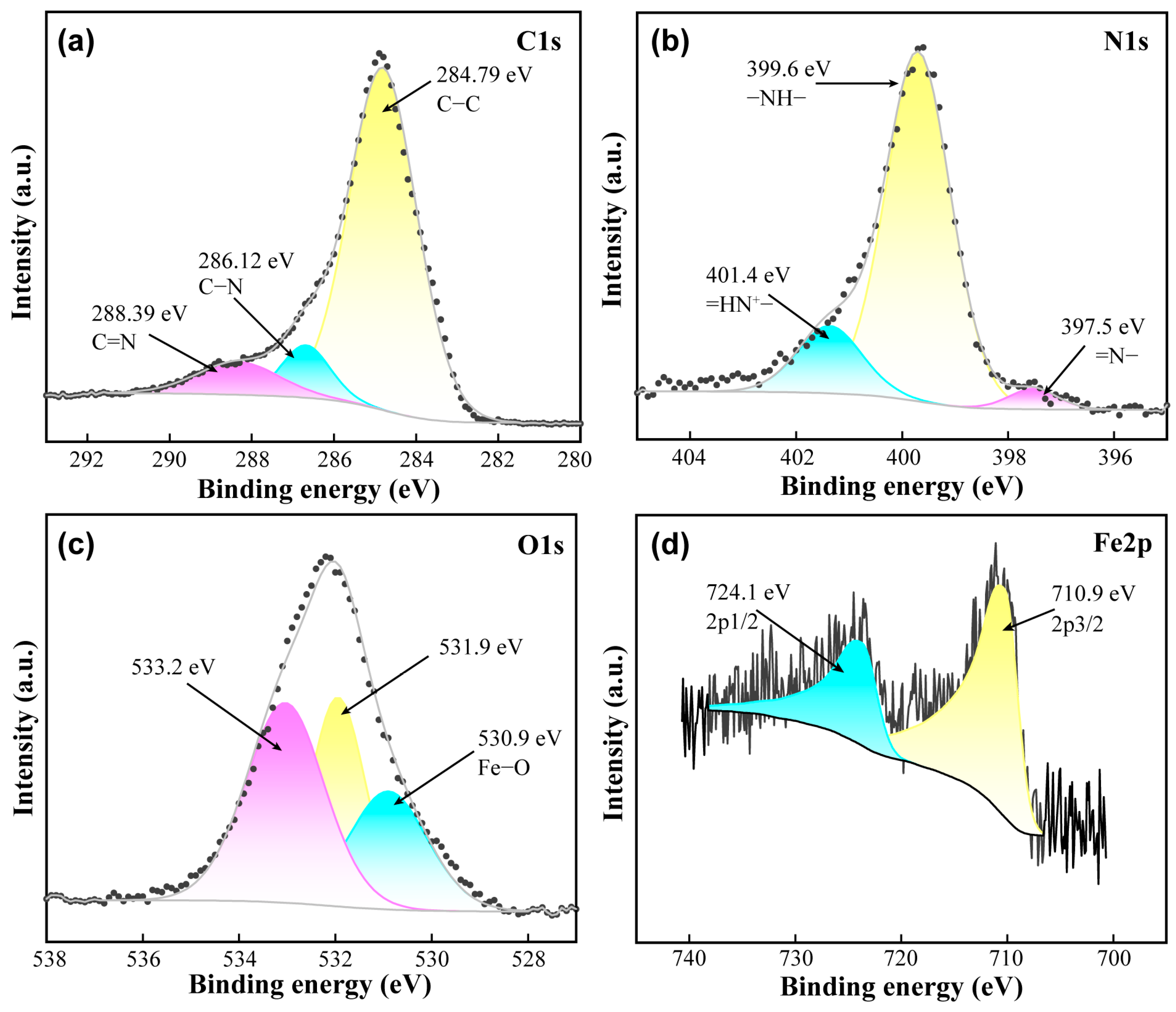
3.1. Characterizations of Samples
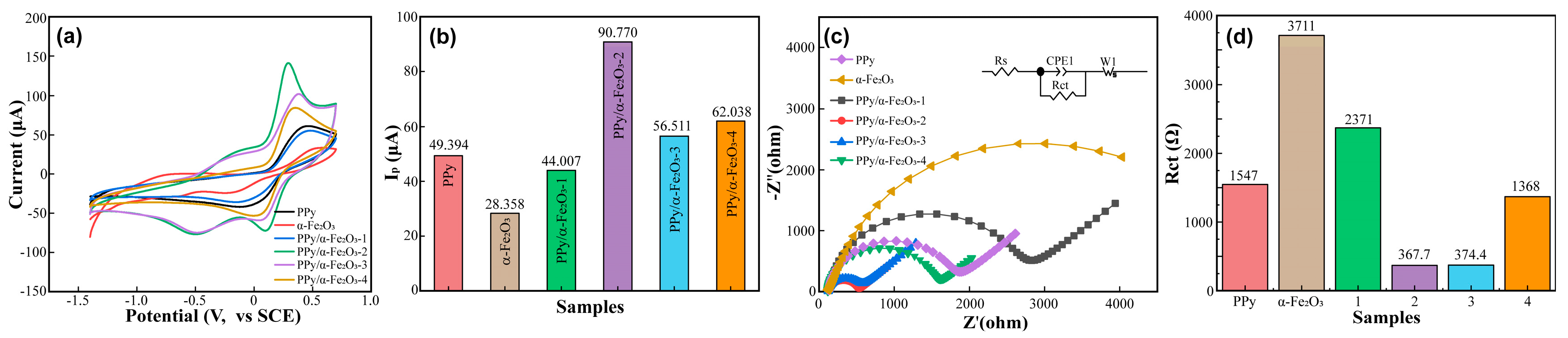
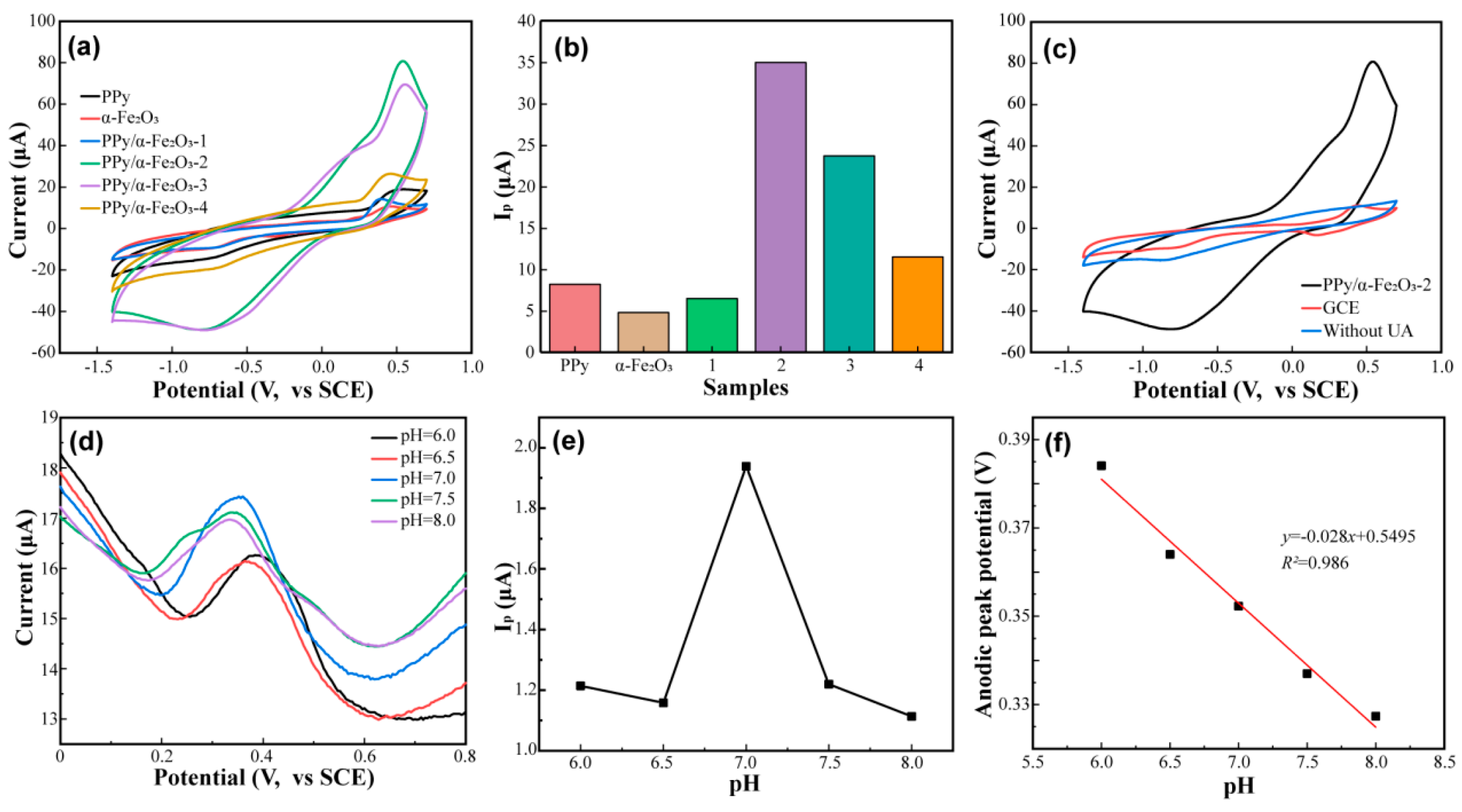
3.2. Electrochemical Performance of PPy/α-Fe2O3 Composite
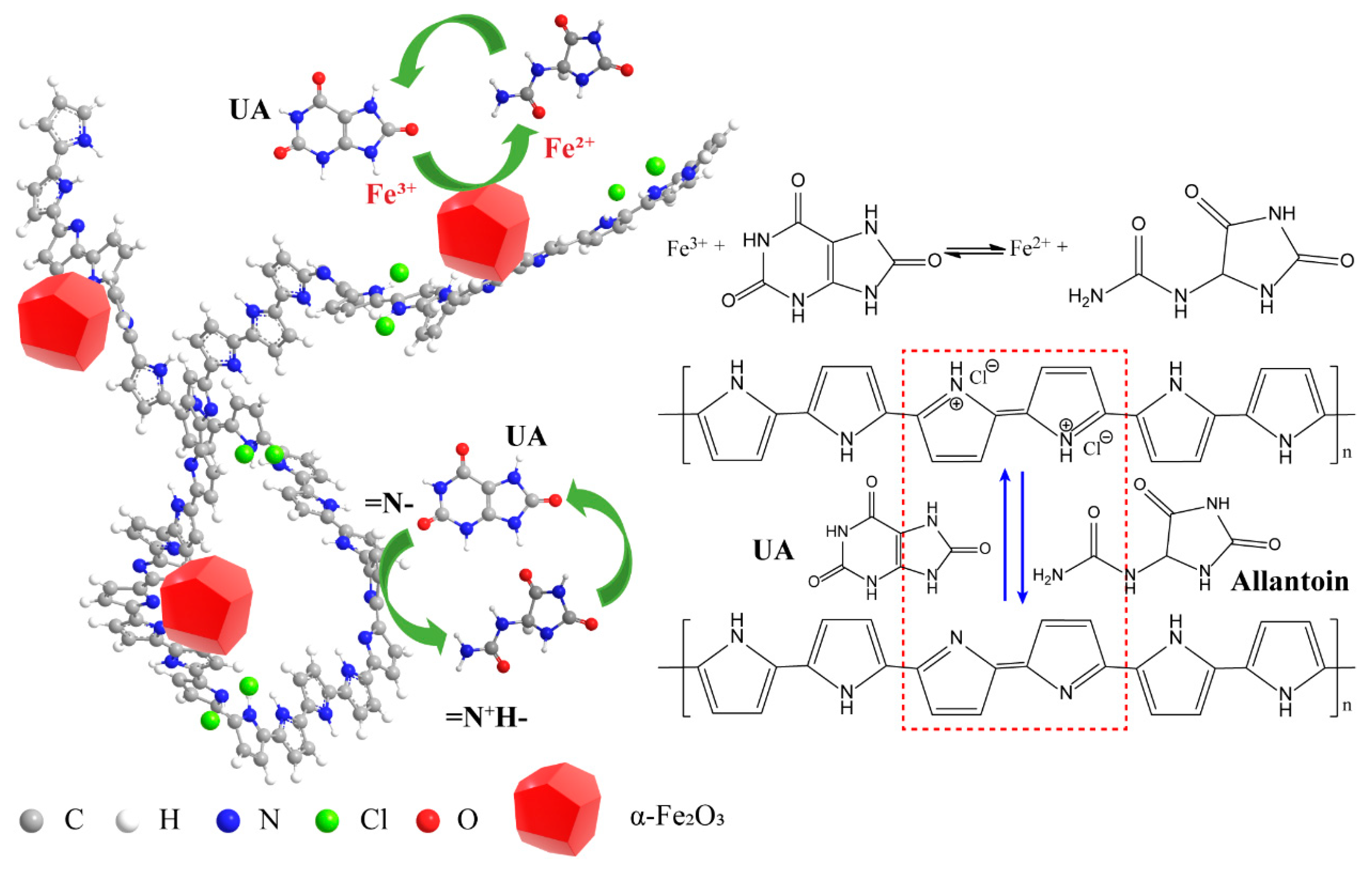
3.3. The Detection Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Y.; Zhou, W.; Wang, Y.; Zhou, R. Gender-Specific Association between Uric Acid Level and Chronic Kidney Disease in the Elderly Health Checkup Population in China. Ren. Fail. 2019, 41, 197–203. [Google Scholar] [CrossRef]
- Motshakeri, M.; Phillips, A.R.J.; Kilmartin, P.A. Application of Cyclic Voltammetry to Analyse Uric Acid and Reducing Agents in Commercial Milks. Food Chem. 2019, 293, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.; Khosravan, R.; Erdmann, C.; Fiene, J.; Lee, J.W. Quantification of Uric Acid, Xanthine and Hypoxanthine in Human Serum by HPLC for Pharmacodynamic Studies. J. Chromatogr. B 2006, 837, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Saadati, A.; Farshchi, F.; Hasanzadeh, M.; Seidi, F. A Microfluidic Paper-Based Colorimetric Device for the Visual Detection of Uric Acid in Human Urine Samples. Anal. Methods 2021, 13, 3909–3921. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yao, Q.; Tang, X.; Zhong, H.; Qiu, P.; Wang, X. A Highly Selective and Sensitive Colorimetric Detection of Uric Acid in Human Serum Based on MoS2-Catalyzed Oxidation TMB. Anal. Bioanal. Chem. 2019, 411, 943–952. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, P.; Deng, X.; Sun, Y.; Wang, X.; Song, D. Enzymatic Determination of Uric Acid Using Water-Soluble CuInS/ZnS Quantum Dots as a Fluorescent Probe. Microchim. Acta 2018, 185, 499. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Kim, Y.I.; Lee, S.W.; Jung, H.G.; Lee, G.; Yoon, D.S. Highly Permselective Uric Acid Detection Using Kidney Cell Membrane–Functionalized Enzymatic Biosensors. Biosens. Bioelectron. 2021, 190, 113411. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Mu, F.; Qin, L.; Jia, X.; Yang, C. Synthesis and Characterization of MgO/ZnO Composite Nanosheets for Biosensor. Mater. Chem. Phys. 2015, 166, 176–181. [Google Scholar] [CrossRef]
- Yan, Q.; Zhi, N.; Yang, L.; Xu, G.; Feng, Q.; Zhang, Q.; Sun, S. A Highly Sensitive Uric Acid Electrochemical Biosensor Based on a Nano-Cube Cuprous Oxide/Ferrocene/Uricase Modified Glassy Carbon Electrode. Sci. Rep. 2020, 10, 10607. [Google Scholar] [CrossRef]
- Zuo, X.; Chang, K.; Zhao, J.; Xie, Z.; Tang, H.; Li, B.; Chang, Z. Bubble-Template-Assisted Synthesis of Hollow Fullerene-like MoS2 Nanocages as a Lithium Ion Battery Anode Material. J. Mater. Chem. A 2016, 4, 51–58. [Google Scholar] [CrossRef]
- Lu, M.; Deng, Y.; Luo, Y.; Lv, J.; Li, T.; Xu, J.; Chen, S.-W.; Wang, J. Graphene Aerogel–Metal–Organic Framework-Based Electrochemical Method for Simultaneous Detection of Multiple Heavy-Metal Ions. Anal. Chem. 2019, 91, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qi, Y.; Shen, Y.; Yuan, Y.; Zhang, L.; Zhang, C.; Sun, Y. A Ratiometric Electrochemical Sensor for Simultaneous Detection of Multiple Heavy Metal Ions Based on Ferrocene-Functionalized Metal-Organic Framework. Sens. Actuators B Chem. 2020, 310, 127756. [Google Scholar] [CrossRef]
- Hu, R.; Zhang, X.; Chi, K.-N.; Yang, T.; Yang, Y.-H. Bifunctional MOFs-Based Ratiometric Electrochemical Sensor for Multiplex Heavy Metal Ions. ACS Appl. Mater. Interfaces 2020, 12, 30770–30778. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, J.; Tong, H.; Huang, Y.; Liu, B.; Yang, X.; Su, Z.; Tu, X.; Qin, X. Simultaneous Electrochemical Detection of Cd (II) and Pb (II) Based on L-Cysteine Functionalized Gold Nanoparticles/Metal-Organic Frameworks-Graphene Oxide Nanocomposites. J. Electroanal. Chem. 2023, 943, 117573. [Google Scholar] [CrossRef]
- Huo, D.; Zhang, Y.; Li, N.; Ma, W.; Liu, H.; Xu, G.; Li, Z.; Yang, M.; Hou, C. Three-Dimensional Graphene/Amino-Functionalized Metal–Organic Framework for Simultaneous Electrochemical Detection of Cd(II), Pb(II), Cu(II), and Hg(II). Anal. Bioanal. Chem. 2022, 414, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Sanna Jilani, B.; Mruthyunjayachari, C.D.; Malathesh, P.; Mounesh; Sharankumar, T.M.; Reddy, K.R.V. Electrochemical Sensing Based MWCNT-Cobalt Tetra Substituted Sorbaamide Phthalocyanine onto the Glassy Carbon Electrode towards the Determination of 2-Amino Phenol: A Voltammetric Study. Sens. Actuators B Chem. 2019, 301, 127078. [Google Scholar] [CrossRef]
- Chaudhary, A.; Khan, M.Q.; Khan, R.A.; Alsalme, A.; Ahmad, K.; Kim, H. Fabrication of CeO2/GCE for Electrochemical Sensing of Hydroquinone. Biosensors 2022, 12, 846. [Google Scholar] [CrossRef]
- Tajik, S.; Beitollahi, H.; Garkani Nejad, F.; Sheikhshoaie, I.; Nugraha, A.S.; Jang, H.W.; Yamauchi, Y.; Shokouhimehr, M. Performance of Metal–Organic Frameworks in the Electrochemical Sensing of Environmental Pollutants. J. Mater. Chem. A 2021, 9, 8195–8220. [Google Scholar] [CrossRef]
- Dong, W.; Wen, H.; Du, X.; Li, L.; Li, Z. Electrochemical Sensing of Tetracycline Based on Au NPs@MoS2/Ch Hybrid Structures. J. Electroanal. Chem. 2022, 923, 116807. [Google Scholar] [CrossRef]
- Mahmood, F.; Sun, Y.; Wan, C. Biomass-Derived Porous Graphene for Electrochemical Sensing of Dopamine. RSC Adv. 2021, 11, 15410–15415. [Google Scholar] [CrossRef] [PubMed]
- Alahmadi, N.; El-Said, W.A. Electrochemical Sensing of Dopamine Using Polypyrrole/Molybdenum Oxide Bilayer-Modified ITO Electrode. Biosensors 2023, 13, 578. [Google Scholar] [CrossRef]
- Zhao, M.; Li, Z.; Zhang, X.; Yu, J.; Ding, Y.; Li, H.; Ma, Y. Employing the Interfacial Barrier of P-rGO/ZnO Microspheres for Improving the Electrochemical Sensing Performance to Dopamine. Sens. Actuators B Chem. 2020, 309, 127757. [Google Scholar] [CrossRef]
- Jalali, M.; Filine, E.; Dalfen, S.; Mahshid, S. Microscale Reactor Embedded with Graphene/Hierarchical Gold Nanostructures for Electrochemical Sensing: Application to the Determination of Dopamine. Microchim. Acta 2020, 187, 90. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Yang, B.; Zhao, Z.; Zhao, Y.; Pan, R.; Wang, D.; Kong, Y.; Chen, Y.; Huang, G.; Kong, J.; et al. Sonication-Triggered Rolling of Janus Porous Nanomembranes for Electrochemical Sensing of Dopamine and Ascorbic Acid. ACS Appl. Nano Mater. 2020, 3, 10032–10039. [Google Scholar] [CrossRef]
- Gao, F.; Yang, Y.; Qiu, W.; Song, Z.; Wang, Q.; Niu, L. Ni 3 C/Ni Nanochains for Electrochemical Sensing of Glucose. ACS Appl. Nano Mater. 2021, 4, 8520–8529. [Google Scholar] [CrossRef]
- Wang, Q.; Jia, Q.; Hu, P.; Ji, L. Tunable Non-Enzymatic Glucose Electrochemical Sensing Based on the Ni/Co Bimetallic MOFs. Molecules 2023, 28, 5649. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Hu, K.; Wang, S.; Chen, X.; Xu, R.; Xiong, X.; Yuan, X.; Zhang, M.; Huang, K. ZIF-8 Derived Ni(OH)2 Hollow Nanocages for Non-Enzymatic Glucose Electrochemical Sensing. J. Mater. Sci. 2022, 57, 18589–18600. [Google Scholar] [CrossRef]
- Saeed, U.; Fatima, B.; Hussain, D.; Imran, M.; Jamil, M.; Naeem Ashiq, M.; Najam-ul-Haq, M. Scalable Synthesis of Ce-MOF Derived CeO/C Hierarchical: Efficient Electrochemical Sensing of Uric Acid as Potential Biomarker in Acute Myeloid Leukaemia Patients. Microchem. J. 2022, 181, 107808. [Google Scholar] [CrossRef]
- Kulyk, B.; Pereira, S.O.; Fernandes, A.J.S.; Fortunato, E.; Costa, F.M.; Santos, N.F. Laser-Induced Graphene from Paper for Non-Enzymatic Uric Acid Electrochemical Sensing in Urine. Carbon 2022, 197, 253–263. [Google Scholar] [CrossRef]
- Hou, T.; Gai, P.; Song, M.; Zhang, S.; Li, F. Synthesis of a Three-Layered SiO2@Au Nanoparticle@polyaniline Nanocomposite and Its Application in Simultaneous Electrochemical Detection of Uric Acid and Ascorbic Acid. J. Mater. Chem. B 2016, 4, 2314–2321. [Google Scholar] [CrossRef]
- Li, G.; Xu, C.; Xu, H.; Gan, L.; Sun, K.; Yuan, B. Tunable Graphene Oxide for the Low-Fouling Electrochemical Sensing of Uric Acid in Human Serum. Analyst 2023, 148, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Eryigit, M.; Kurt Urhan, B.; Dogan, H.O.; Ozer, T.O.; Demir, U. ZnO Nanosheets-Decorated ERGO Layers: An Efficient Electrochemical Sensor for Non-Enzymatic Uric Acid Detection. IEEE Sens. J. 2022, 22, 5555–5561. [Google Scholar] [CrossRef]
- Ibrahim, A.A.; Kumar, R.; Umar, A.; Kim, S.H.; Bumajdad, A.; Ansari, Z.A.; Baskoutas, S. Cauliflower-Shaped ZnO Nanomaterials for Electrochemical Sensing and Photocatalytic Applications. Electrochim. Acta 2016, 222, 463–472. [Google Scholar] [CrossRef]
- Zhuang, Z.; Chen, Y.; Chen, K.; Liu, Z.; Guo, Z.; Huang, X. In-Situ Synthesis of ZnO onto Nanoporous Gold Microelectrode for the Electrochemical Sensing of Arsenic(III) in near-Neutral Conditions. Sens. Actuators B Chem. 2023, 378, 133184. [Google Scholar] [CrossRef]
- Sundar, S.; Venkatachalam, G.; Kwon, S. Biosynthesis of Copper Oxide (CuO) Nanowires and Their Use for the Electrochemical Sensing of Dopamine. Nanomaterials 2018, 8, 823. [Google Scholar] [CrossRef]
- Manigandan, R.; Dhanasekaran, T.; Padmanaban, A.; Giribabu, K.; Suresh, R.; Narayanan, V. Bifunctional Hexagonal Ni/NiO Nanostructures: Influence of the Core–Shell Phase on Magnetism, Electrochemical Sensing of Serotonin, and Catalytic Reduction of 4-Nitrophenol. Nanoscale Adv. 2019, 1, 1531–1540. [Google Scholar] [CrossRef]
- Hao, C.; Shen, Y.; Wang, Z.; Wang, X.; Feng, F.; Ge, C.; Zhao, Y.; Wang, K. Preparation and Characterization of Fe2O3 Nanoparticles by Solid-Phase Method and Its Hydrogen Peroxide Sensing Properties. ACS Sustain. Chem. Eng. 2016, 4, 1069–1077. [Google Scholar] [CrossRef]
- Wu, S.; Xiong, W.; Li, H. Insights into the Fe Oxidation State of Sphere-like Fe2O3 Nanoparticles for Simultaneous Pb2+ and Cu2+ Detection. J. Alloys Compd. 2023, 934, 167863. [Google Scholar] [CrossRef]
- Immanuel, S.; Aparna, T.K.; Sivasubramanian, R. A Facile Preparation of Au—SiO2 Nanocomposite for Simultaneous Electrochemical Detection of Dopamine and Uric Acid. Surf. Interfaces 2019, 14, 82–91. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Kang, P.; Wang, S.-Q.; Liu, Z.-G.; Li, Y.-X.; Guo, Z. Ag Nanoparticles Anchored onto Porous CuO Nanobelts for the Ultrasensitive Electrochemical Detection of Dopamine in Human Serum. Sens. Actuators B Chem. 2021, 327, 128878. [Google Scholar] [CrossRef]
- He, W.; Ye, X.; Cui, T. Flexible Electrochemical Sensor with Graphene and Gold Nanoparticles to Detect Dopamine and Uric Acid. IEEE Sens. J. 2021, 21, 26556–26565. [Google Scholar] [CrossRef]
- Priya, C.; Anuja, S.; Babu, R.S.; Narayanan, S.S. Low Potential Determination of Dopamine in the Presence of Ascorbic Acid Using Phenothiazine Dye–Functionalized Graphite Oxide–Modified Electrode. Ionics 2023, 29, 5481–5490. [Google Scholar] [CrossRef]
- Husain, A.; Al-Zahrani, S.A.; Al Otaibi, A.; Khan, I.; Mujahid Ali Khan, M.; Alosaimi, A.M.; Khan, A.; Hussein, M.A.; Asiri, A.M.; Jawaid, M. Fabrication of Reproducible and Selective Ammonia Vapor Sensor-Pellet of Polypyrrole/Cerium Oxide Nanocomposite for Prompt Detection at Room Temperature. Polymers 2021, 13, 1829. [Google Scholar] [CrossRef]
- Lett, J.A.; Sagadevan, S.; Alshahateet, S.F.; Murugan, B.; Jasni, A.H.; Fatimah, I.; Hossain, M.A.M.; Mohammad, F.; Oh, W.C. Synthesis and Characterization of Polypyrrole-Coated Iron Oxide Nanoparticles. Mater. Res. Express 2021, 8, 025007. [Google Scholar] [CrossRef]
- Morsy, R.; Hosny, M.; Reicha, F.; Elnimr, T. Development and Characterization of Multifunctional Electrospun Ferric Oxide-Gelatin-Glycerol Nanofibrous Mat for Wound Dressing Applications. Fibers Polym. 2016, 17, 2014–2019. [Google Scholar] [CrossRef]
- Beniwal, A.; Sunny. Novel TPU/Fe2O3 and TPU/Fe2O3/PPy Nanocomposites Synthesized Using Electrospun Nanofibers Investigated for Analyte Sensing Applications at Room Temperature. Sens. Actuators B Chem. 2020, 304, 127384. [Google Scholar] [CrossRef]
- Wang, C.; Yang, M.; Liu, L.; Xu, Y.; Zhang, X.; Cheng, X.; Gao, S.; Gao, Y.; Huo, L. One-Step Synthesis of Polypyrrole/Fe2O3 Nanocomposite and the Enhanced Response of NO2 at Low Temperature. J. Colloid. Interface Sci. 2020, 560, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-L.; Guo, J.-W.; Wang, Y.-W.; Wang, A.-Z.; Yu, X.; Ding, L.-H. A Flexible Electrochemical Sensor for Paracetamol Based on Porous Honeycomb-like NiCo-MOF Nanosheets. Rare Met. 2023, 42, 3311–3317. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, G.; Shi, Q.; Zhao, R.; Chen, M. Preparation of Thiolated Calix [8]Arene/AuNPs/MWCNTs Modified Glassy Carbon Electrode and Its Electrocatalytic Oxidation toward Paracetamol. Sens. Actuators B Chem. 2018, 277, 289–296. [Google Scholar] [CrossRef]
- Abrori, S.A.; Septiani, N.L.W.; Nugraha; Nuruddin, A.; Anshori, I.; Yuliarto, B. Comparison of a 2D/3D Imidazole-Based MOF and Its Application as a Non-Enzymatic Electrochemical Sensor for the Detection of Uric Acid. New J. Chem. 2022, 46, 21342–21349. [Google Scholar] [CrossRef]
- Li, F.; Liu, L.; Liu, T.; Zhang, M. Ni-MOF Nanocomposites Decorated by Au Nanoparticles: An Electrochemical Sensor for Detection of Uric Acid. Ionics 2022, 28, 4843–4851. [Google Scholar] [CrossRef]
- Dutta, P.; Sharma, V.; Bhardwaj, H.; Agrawal, V.V.; Rajesh; Sumana, G. Fabrication of Electrochemical Biosensor Using Zinc Oxide Nanoflowers for the Detection of Uric Acid. MAPAN 2022, 37, 585–595. [Google Scholar] [CrossRef]
- Babu, R.S.; Prabhu, P.; Narayanan, S.S. Selective Electrooxidation of Uric Acid in Presence of Ascorbic Acid at a Room Temperature Ionic Liquid/Nickel Hexacyanoferarrate Nanoparticles Composite Electrode. Colloids Surf. B Biointerfaces 2011, 88, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Ipekci, H.H. Holey MoS2-Based Electrochemical Sensors for Simultaneous Dopamine and Uric Acid Detection. Anal. Methods 2023, 15, 2989–2996. [Google Scholar] [CrossRef]
- John, J.F.; Dhinasekaran, D.; Subashchandran, S. Cobalt Doped NiFe2O4 on 3D Nickel Foam Substrate for Electrochemical Detection of Uric Acid. Surf. Interfaces 2024, 44, 103629. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Detection Range | LOD | LOQ | References |
|---|---|---|---|---|
| W-ZIF-67 | 20–1000 μM | 3.04 μM | 9.12 μM | [50] |
| Au/Ni-MOF | 10–500 μM | 5.6 μM | 16.8 μM | [51] |
| UOx/ZnONFs | 5–750 μM | 130 μM | 390 μM | [52] |
| RTIL-NiHCF-NP-Gel | 1.0–2600.0 μM | 0.33 μM | 1 μM | [53] |
| GP5AuNPs5 | 20–500 μM | 1.47 μM | 4.41 μM | [41] |
| Holey MoS2 | 200–700 μM | 5.62 μM | 16.86 μM | [54] |
| Co0.01Ni0.99Fe2O4 | 4–5280 μM | 6.38 μM | 19.14 μM | [55] |
| PPy/Fe2O3 | 5–200 μM | 1.349 μM | 4.407 μM | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Liu, S.; Song, X.; Jiang, K.; Hou, Y.; Cheng, Q.; Miao, W.; Tian, L.; Ren, Y.; Xu, S. Polypyrrole/α-Fe2O3 Hybrids for Enhanced Electrochemical Sensing Performance towards Uric Acid. Coatings 2024, 14, 227. https://doi.org/10.3390/coatings14020227
Wang R, Liu S, Song X, Jiang K, Hou Y, Cheng Q, Miao W, Tian L, Ren Y, Xu S. Polypyrrole/α-Fe2O3 Hybrids for Enhanced Electrochemical Sensing Performance towards Uric Acid. Coatings. 2024; 14(2):227. https://doi.org/10.3390/coatings14020227
Chicago/Turabian StyleWang, Renjie, Shanshan Liu, Xudong Song, Kai Jiang, Yaohui Hou, Qiaohuan Cheng, Wei Miao, Li Tian, Ying Ren, and Sankui Xu. 2024. "Polypyrrole/α-Fe2O3 Hybrids for Enhanced Electrochemical Sensing Performance towards Uric Acid" Coatings 14, no. 2: 227. https://doi.org/10.3390/coatings14020227
APA StyleWang, R., Liu, S., Song, X., Jiang, K., Hou, Y., Cheng, Q., Miao, W., Tian, L., Ren, Y., & Xu, S. (2024). Polypyrrole/α-Fe2O3 Hybrids for Enhanced Electrochemical Sensing Performance towards Uric Acid. Coatings, 14(2), 227. https://doi.org/10.3390/coatings14020227