
Systematic First-Principles Investigations of the Nucleation, Growth, and Surface Properties of Al11RE3 Second-Phase Particles in Al-Based Alloys

Abstract
:1. Introduction
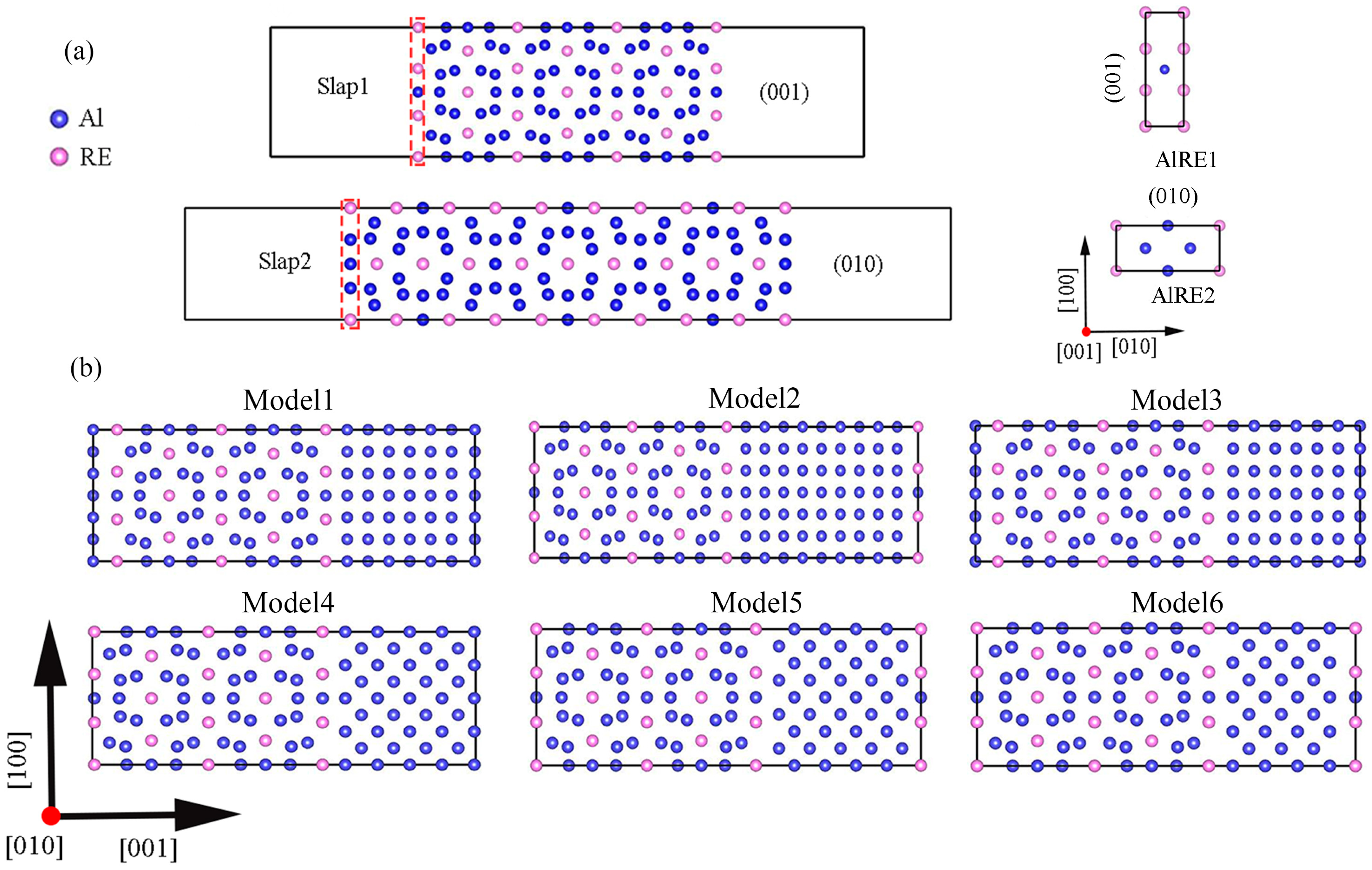
2. Computational Methodology
3. Result and Discussion
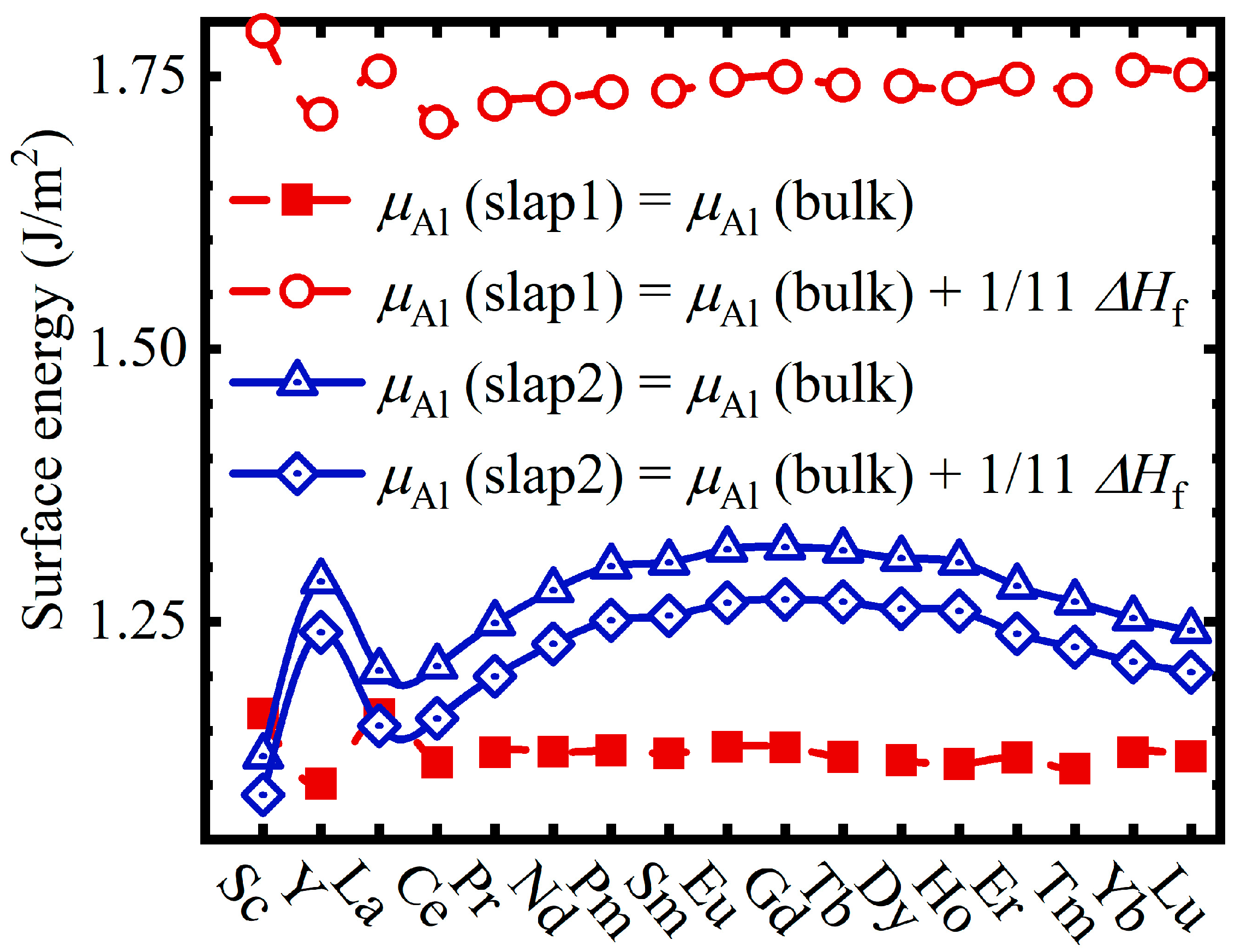
3.1. Surface Activity
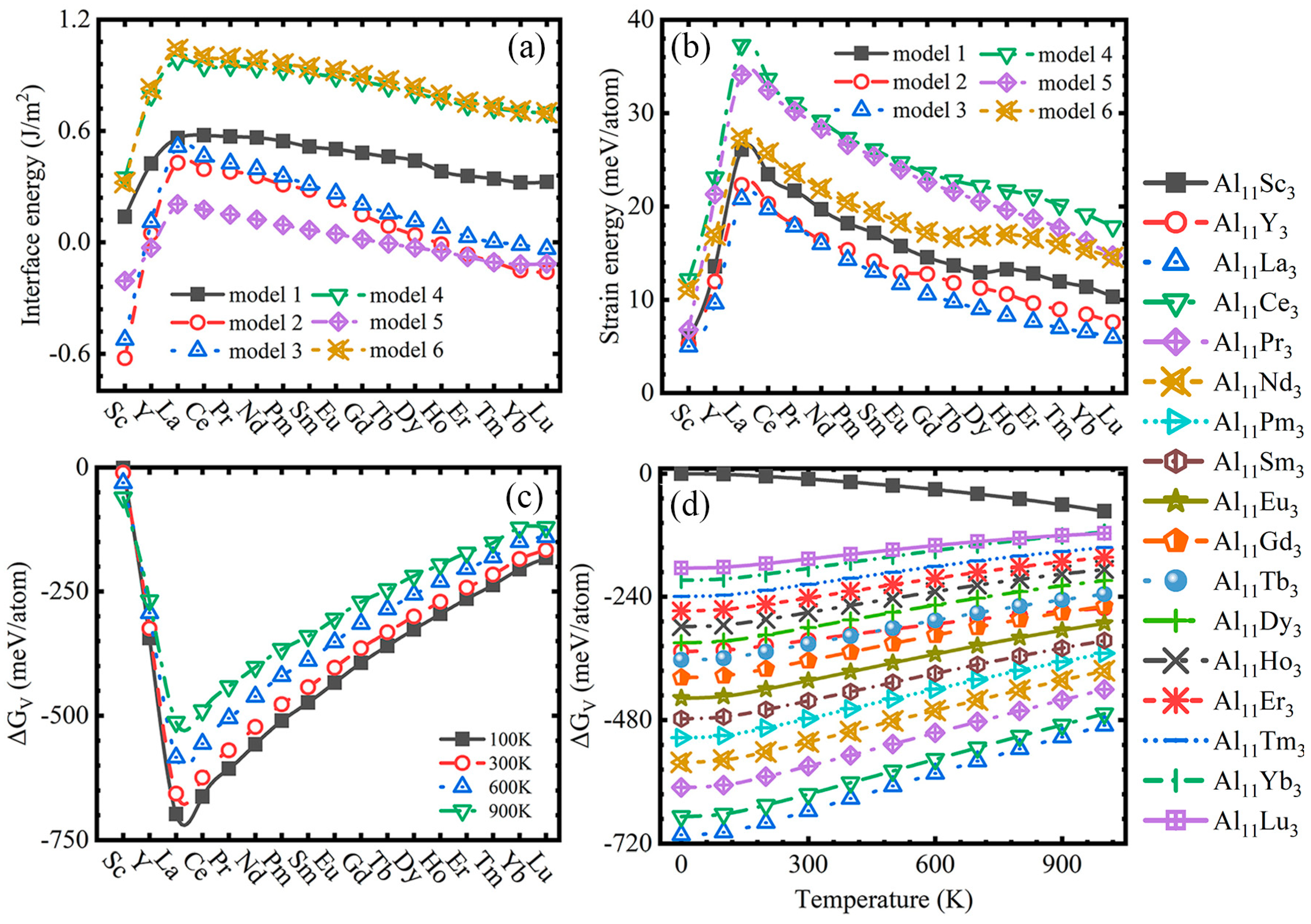
3.2. Driving and Hindering Forces
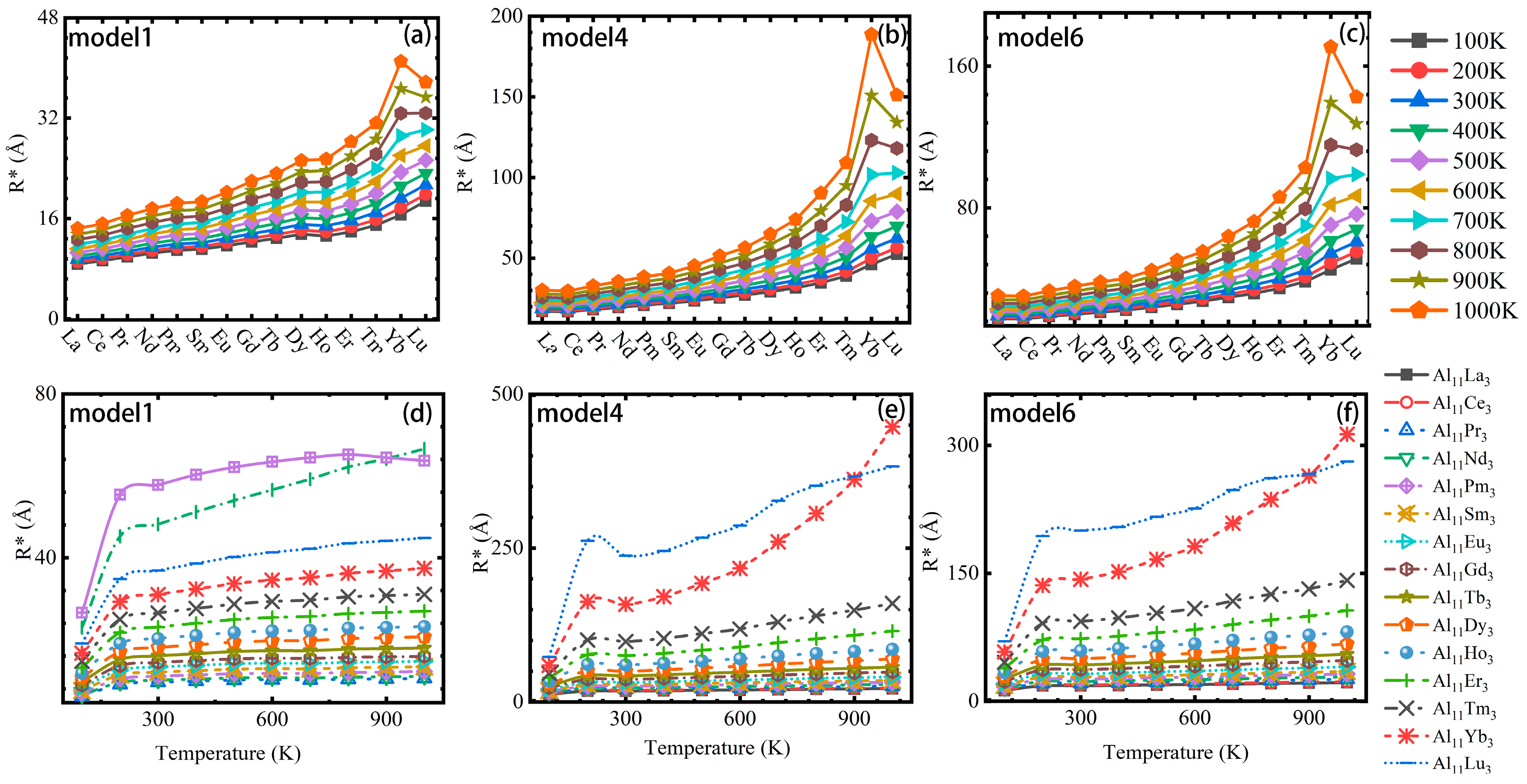
3.3. Particle Nucleation and Growth
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marquis, E.; Seidman, D.N. Nanoscale structural evolution of Al3Sc precipitates in Al (Sc) alloys. Acta Mater. 2001, 49, 1909–1919. [Google Scholar] [CrossRef]
- Woodward, C.; Asta, M.; Kresse, G.; Hafner, J. Density of constitutional and thermal point defects in L12 Al3Sc. Phys. Rev. B 2001, 63, 094103. [Google Scholar] [CrossRef]
- Clouet, E.; Nastar, M.; Sigli, C. Nucleation of Al3Zr and Al3Sc in aluminum alloys: From kinetic Monte Carlo simulations to classical theory. Phys. Rev. B 2004, 69, 064109. [Google Scholar] [CrossRef]
- Fu, L.; Ke, J.L.; Zhang, Q.; Tang, B.Y.; Peng, L.M.; Ding, W.J. Mechanical properties of L12 type Al3X (X = Mg, Sc, Zr) from first-principles study. Phys. Status Solidi 2012, 249, 1510–1516. [Google Scholar] [CrossRef]
- Choe, H.; Dunand, D.C. Synthesis, structure, and mechanical properties of Ni–Al and Ni–Cr–Al superalloy foams. Acta Mater. 2004, 52, 1283–1295. [Google Scholar] [CrossRef]
- Nong, Z.-S.; Zhu, J.-C.; Yang, X.-W.; Cao, Y.; Lai, Z.-H.; Liu, Y. First-principles investigation of the elastic and electronic properties of the binary intermetallics in the Al–La alloy system. Phys. B Condens. Matter 2012, 407, 4706–4711. [Google Scholar] [CrossRef]
- Yao, F.; Zhang, L.; Meng, J.; Liu, X.; Meng, J.; Zhang, H. Strategy to enhance intrinsic ductility and surface stability of Mg-Zn-R (R = La to Sm) alloys from a first-principles study. Comput. Mater. Sci. 2019, 165, 96–100. [Google Scholar] [CrossRef]
- Azzeddine, H.; Hanna, A.; Dakhouche, A.; Rabahi, L.; Scharnagl, N.; Dopita, M.; Brisset, F.; Helbert, A.-L.; Baudin, T. Impact of rare-earth elements on the corrosion performance of binary magnesium alloys. J. Alloys Compd. 2020, 829, 154569. [Google Scholar] [CrossRef]
- Tolley, A.; Radmilovic, V.; Dahmen, U. Segregation in Al3 (Sc, Zr) precipitates in Al–Sc–Zr alloys. Scr. Mater. 2005, 52, 621–625. [Google Scholar] [CrossRef]
- Li, J.; Oberdorfer, B.; Wurster, S.; Schumacher, P. Impurity effects on the nucleation and growth of primary Al3 (Sc, Zr) phase in Al alloys. J. Mater. Sci. 2014, 49, 5961–5977. [Google Scholar] [CrossRef]
- Hyde, K.; Norman, A.; Prangnell, P.B. The effect of Ti on grain refinement in Al-Sc alloys. In Proceedings of the Materials Science Forum, Cambridge, UK, 2–5 July 2002; pp. 39–44. [Google Scholar]
- Nie, Z.R.; Jin, T.; Fu, J.; Xu, G.; Yang, J.; Zhou, J.X.; Zuo, T.Y. Research on rare earth in aluminum. In Materials Science Forum; Trans Tech Publications Ltd.: Zurich, Switzerland, 2002; p. 1731. [Google Scholar]
- Meng, L.; Zheng, X. Overview of the effects of impurities and rare earth elements in Al-Li alloys. Mater. Sci. Eng. A 1997, 237, 109–118. [Google Scholar] [CrossRef]
- Shi, Y.; Pan, Q.; Li, M.; Huang, X.; Li, B. Effect of Sc and Zr additions on corrosion behaviour of Al–Zn–Mg–Cu alloys. J. Alloys Compd. 2014, 612, 42–50. [Google Scholar] [CrossRef]
- Murashkin, M.Y.; Sabirov, I.; Medvedev, A.; Enikeev, N.; Lefebvre, W.; Valiev, R.; Sauvage, X. Mechanical and electrical properties of an ultrafine grained Al–8.5 wt.% RE (RE = 5.4 wt.% Ce, 3.1 wt.% La) alloy processed by severe plastic deformation. Mater. Des. 2016, 90, 433–442. [Google Scholar] [CrossRef]
- Pérez-Bustamante, R.; Reyna-Cruz, A.; Acosta-Peña, D.; Santillán-Rodríguez, C.; Matutes-Aquino, J.; Pérez-Bustamante, F.; Maldonado-Orozco, M.; Aguilar-Santillán, J.; Martínez-Sánchez, R. Effect of cerium/lanthanum addition on microstructure and mechanical properties of Al7075 alloy via mechanical alloying and sintering. J. Rare Earths 2016, 34, 420–427. [Google Scholar] [CrossRef]
- Srivastava, V.C.; Surreddi, K.B.; Scudino, S.; Schowalter, M.; Uhlenwinkel, V.; Schulz, A.; Eckert, J.; Rosenauer, A.; Zoch, H.-W. Microstructural characteristics of spray formed and heat treated Al–(Y, La)–Ni–Co system. J. Alloys Compd. 2013, 578, 471–480. [Google Scholar] [CrossRef]
- Amadeh, A.; Pahlevani, B.; Heshmati-Manesh, S. Effects of rare earth metal addition on surface morphology and corrosion resistance of hot-dipped zinc coatings. Corros. Sci. 2002, 44, 2321–2331. [Google Scholar] [CrossRef]
- Rosalbino, F.; Angelini, E.; Macciò, D.; Saccone, A.; Delfino, S. Application of EIS to assess the effect of rare earths small addition on the corrosion behaviour of Zn–5% Al (Galfan) alloy in neutral aerated sodium chloride solution. Electrochim. Acta 2009, 54, 1204–1209. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S. First-principles study of thermodynamic properties and solubility of aluminum-rare-earth intermetallics. Comput. Mater. Sci. 2014, 90, 56–60. [Google Scholar] [CrossRef]
- Mao, Z.; Seidman, D.N.; Wolverton, C. First-principles phase stability, magnetic properties and solubility in aluminum–rare-earth (Al–RE) alloys and compounds. Acta Mater. 2011, 59, 3659–3666. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, B.; Li, X.; Li, Q.; Ye, T.; Xiang, S.; Zhao, M.-C.; Atrens, A. In-situ EBSD study on twinning activity caused by deep cryogenic treatment (DCT) for an as-cast AZ31 Mg alloy. J. Mater. Res. Technol. 2024, 30, 3840–3850. [Google Scholar] [CrossRef]
- Vlach, M.; Čížek, J.; Smola, B.; Melikhova, O.; Vlček, M.; Kodetová, V.; Kudrnová, H.; Hruška, P. Heat treatment and age hardening of Al–Si–Mg–Mn commercial alloy with addition of Sc and Zr. Mater. Charact. 2017, 129, 1–8. [Google Scholar] [CrossRef]
- Murty, B.; Kori, S.; Chakraborty, M. Grain refinement of aluminium and its alloys by heterogeneous nucleation and alloying. Int. Mater. Rev. 2002, 47, 3–29. [Google Scholar] [CrossRef]
- Krug, M.E.; Werber, A.; Dunand, D.C.; Seidman, D.N. Core–shell nanoscale precipitates in Al–0.06 at.% Sc microalloyed with Tb, Ho, Tm or Lu. Acta Mater. 2010, 58, 134–145. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, G.; Sun, J. Recent progress in high-temperature resistant aluminum-based alloys: Microstructural design and precipitation strategy. Acta Metall. Sin. 2020, 57, 129–149. [Google Scholar]
- Weakley-Bollin, S.; Donlon, W.; Donlon, W.; Wolverton, C.; Allison, J.; Jones, J.W. Modeling the age-hardening behavior of Al-Si-Cu alloys. Metall. Mater. Trans. A 2004, 35, 2407–2418. [Google Scholar] [CrossRef]
- Polmear, I.; Couper, M.J. Design and development of an experimental wrought aluminum alloy for use at elevated temperatures. Metall. Trans. A 1988, 19, 1027–1035. [Google Scholar] [CrossRef]
- Yang, J.; Heogh, W.; Ju, H.; Kang, S.; Jang, T.-S.; Jung, H.-D.; Jahazi, M.; Han, S.C.; Park, S.J.; Kim, H.S. Functionally graded structure of a nitride-strengthened Mg2Si-based hybrid composite. J. Magnes. Alloys 2024, 12, 1239–1256. [Google Scholar] [CrossRef]
- Suwanpreecha, C.; Toinin, J.P.; Michi, R.; Pandee, P.; Dunand, D.C.; Limmaneevichitr, C. Strengthening mechanisms in AlNiSc alloys containing Al3Ni microfibers and Al3Sc nanoprecipitates. Acta Mater. 2019, 164, 334–346. [Google Scholar] [CrossRef]
- Pandey, P.; Makineni, S.K.; Gault, B.; Chattopadhyay, K. On the origin of a remarkable increase in the strength and stability of an Al rich Al-Ni eutectic alloy by Zr addition. Acta Mater. 2019, 170, 205–217. [Google Scholar] [CrossRef]
- Suwanpreecha, C.; Pandee, P.; Patakham, U.; Limmaneevichitr, C. New generation of eutectic Al-Ni casting alloys for elevated temperature services. Mater. Sci. Eng. A 2018, 709, 46–54. [Google Scholar] [CrossRef]
- Plotkowski, A.; Rios, O.; Sridharan, N.; Sims, Z.; Unocic, K.; Ott, R.; Dehoff, R.; Babu, S.J.A.M. Evaluation of an Al-Ce alloy for laser additive manufacturing. Acta Mater. 2017, 126, 507–519. [Google Scholar] [CrossRef]
- Sims, Z.C.; Rios, O.R.; Weiss, D.; Turchi, P.E.; Perron, A.; Lee, J.R.; Li, T.T.; Hammons, J.A.; Bagge-Hansen, M.; Willey, T.M. High performance aluminum–cerium alloys for high-temperature applications. Mater. Horiz. 2017, 4, 1070–1078. [Google Scholar] [CrossRef]
- Liu, Y.; Michi, R.A.; Dunand, D.C. Cast near-eutectic Al-12.5 wt.% Ce alloy with high coarsening and creep resistance. Mater. Sci. Eng. A 2019, 767, 138440. [Google Scholar] [CrossRef]
- Sun, Y.; Hung, C.; Hebert, R.J.; Fennessy, C.; Tulyani, S.; Aindow, M. Eutectic microstructures in dilute Al-Ce and Al-Co alloys. Mater. Charact. 2019, 154, 269–276. [Google Scholar] [CrossRef]
- Seidman, D.N.; Marquis, E.A.; Dunand, D.C. Precipitation strengthening at ambient and elevated temperatures of heat-treatable Al (Sc) alloys. Acta Mater. 2002, 50, 4021–4035. [Google Scholar] [CrossRef]
- Booth-Morrison, C.; Mao, Z.; Diaz, M.; Dunand, D.; Wolverton, C.; Seidman, D. Role of silicon in accelerating the nucleation of Al3 (Sc, Zr) precipitates in dilute Al–Sc–Zr alloys. Acta Mater. 2012, 60, 4740–4752. [Google Scholar] [CrossRef]
- Vo, N.; Dunand, D.; Seidman, D. Improving aging and creep resistance in a dilute Al–Sc alloy by microalloying with Si, Zr and Er. Acta Mater. 2014, 63, 73–85. [Google Scholar] [CrossRef]
- Vo, N.Q.; Dunand, D.C.; Seidman, D.N. Role of silicon in the precipitation kinetics of dilute Al-Sc-Er-Zr alloys. Mater. Sci. Eng. A 2016, 677, 485–495. [Google Scholar] [CrossRef]
- Michi, R.A.; Toinin, J.P.; Farkoosh, A.R.; Seidman, D.N.; Dunand, D.C. Effects of Zn and Cr additions on precipitation and creep behavior of a dilute Al–Zr–Er–Si alloy. Acta Mater. 2019, 181, 249–261. [Google Scholar] [CrossRef]
- Booth-Morrison, C.; Dunand, D.C.; Seidman, D.N. Coarsening resistance at 400 C of precipitation-strengthened Al–Zr–Sc–Er alloys. Acta Mater. 2011, 59, 7029–7042. [Google Scholar] [CrossRef]
- Booth-Morrison, C.; Seidman, D.N.; Dunand, D.C. Effect of Er additions on ambient and high-temperature strength of precipitation-strengthened Al–Zr–Sc–Si alloys. Acta Mater. 2012, 60, 3643–3654. [Google Scholar] [CrossRef]
- De Luca, A.; Seidman, D.N.; Dunand, D.C. Effects of Mo and Mn microadditions on strengthening and over-aging resistance of nanoprecipitation-strengthened Al-Zr-Sc-Er-Si alloys. Acta Mater. 2019, 165, 1–14. [Google Scholar] [CrossRef]
- Vo, N.Q.; Seidman, D.N.; Dunand, D.C. Effect of Si micro-addition on creep resistance of a dilute Al-Sc-Zr-Er alloy. Mater. Sci. Eng. A 2018, 734, 27–33. [Google Scholar] [CrossRef]
- De Luca, A.; Seidman, D.N.; Dunand, D.C. Mn and Mo additions to a dilute Al-Zr-Sc-Er-Si-based alloy to improve creep resistance through solid-solution-and precipitation-strengthening. Acta Mater. 2020, 194, 60–67. [Google Scholar] [CrossRef]
- Nie, Z.R.; Wen, S.P.; Huang, H.; Li, B.L.; Zuo, T.Y. Research progress of er-containing aluminum alloy. Chin. J. Nonferrous Metals 2011, 21, 2361–2370. [Google Scholar]
- Lv, S.; Li, Y.; Meng, F.; Duan, Q.; Yang, Q.; Liu, X.; Meng, J.J.C.M.S. Thermodynamic stability of Al11RE3 intermetallic compounds from first-principles calculations. Comput. Mater. Sci. 2017, 131, 28–34. [Google Scholar] [CrossRef]
- Krug, M.E.; Dunand, D.C. Modeling the creep threshold stress due to climb of a dislocation in the stress field of a misfitting precipitate. Acta Mater. 2011, 59, 5125–5134. [Google Scholar] [CrossRef]
- Buch, F.v.; Schumann, S.; Aghion, E.; Bronfin, B.; Mordike, B.; Bamberger, M.; Eliezer, D. Development of a Low-cost, Temperature-and Creep-resistant Magnesium Die-casting Alloy. Magnes. Alloys Their Appl. 2000, 23–28. [Google Scholar] [CrossRef]
- Rzychoń, T.; Kiełbus, A.; Lityńska-Dobrzyńska, L. Microstructure, microstructural stability and mechanical properties of sand-cast Mg–4Al–4RE alloy. Mater. Charact. 2013, 83, 21–34. [Google Scholar] [CrossRef]
- Hu, T.; Ruan, Z.; Fan, T.; Wang, K.; He, K.; Wu, Y. First-principles investigation of the diffusion of TM and the nucleation and growth of L12 Al3TM particles in Al alloys. Crystals 2023, 13, 1032. [Google Scholar] [CrossRef]
- Hu, T.; Ruan, Z.; Fan, T.; Chen, D.; Wu, Y.; Tang, P. First-principles calculations to investigate stability, mechanical and thermo-dynamic properties of AlxTMy intermetallics in aluminum alloys. Solid State Commun. 2023, 376, 115361. [Google Scholar] [CrossRef]
- Fan, T.; Lin, L.; Liang, H.; Ma, Y.; Tang, Y.; Hu, T.; Ruan, Z.; Chen, D.; Wu, Y. First-principles study of the structural, mechanical and thermodynamic properties of Al11RE3 in aluminum alloys. Crystals 2023, 13, 347. [Google Scholar] [CrossRef]
- Fu, J.; Sun, J.; Cen, X.; Zhang, X.; Li, F.; Wu, Y. Growth behavior and orientation relationships in AISI 304 stainless steel during directional solidification. Mater. Charact. 2018, 139, 241–248. [Google Scholar] [CrossRef]
- Hta, B.; Rui, Z.B.; Dong, Q.C.; Xin, L.B.; Qiu, F. Interfacial reaction behavior and evolution mechanism at a preoxidized SiCox/Al interface. J. Mater. Res. Technol. 2021, 15, 1100–1114. [Google Scholar]
- Marder, M. Correlations and Ostwald ripening. Phys. Rev. A 1987, 36, 858. [Google Scholar] [CrossRef] [PubMed]
- Czerwinski, F. Thermal Stability of Aluminum Alloys. Materials 2020, 13, 3441. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, X.; Wang, H.; Jiang, H.; Lei, W.; Jiang, Y.; Yi, D. First-principles investigations on the electronic properties and stabilities of low-index surfaces of L12-Al3Sc intermetallic. Appl. Surf. Sci. 2014, 288, 609–618. [Google Scholar] [CrossRef]
- Mantl, S.; Petry, W.; Schroeder, K.; Vogl, G. Diffusion of iron in aluminum studied by Mössbauer spectroscopy. Phys. Rev. B 1983, 27, 5313. [Google Scholar] [CrossRef]
- Mao, Z.; Chen, W.; Seidman, D.N.; Wolverton, C. First-principles study of the nucleation and stability of ordered precipitates in ternary Al-Sc-Li alloys. Acta Mater. 2011, 59, 3012–3023. [Google Scholar] [CrossRef]
- Hood, G.M. The diffusion of iron in aluminium. Philos. Mag. 1970, 21, 305–328. [Google Scholar] [CrossRef]
- Finnis, M.W. REVIEW ARTICLE: The theory of metal-ceramic interfaces. J. Phys. Condens. Matter 1996, 8, 5811–5836. [Google Scholar] [CrossRef]
- Wang, C.; Wang, C.Y. Ni/Ni3Al interface: A density functional theory study. Appl. Surf. Sci. 2009, 255, 3669–3675. [Google Scholar] [CrossRef]
- Zhou, W.F.; Ren, X.D.; Ren, Y.P.; Yuan, S.Q.; Ren, N.F.; Yang, X.Q.; Adu-Gyamfi, S. Initial dislocation density effect on strain hardening in FCC aluminium alloy under laser shock peening. Philos. Mag. 2017, 97, 1285073. [Google Scholar] [CrossRef]
- Zhao, S.J.; Stocks, G.M.; Zhang, Y.W. Stacking fault energies of face-centered cubic concentrated solid solution alloys. Acta Mater. 2017, 134, 334–345. [Google Scholar] [CrossRef]
- Dong, T.H.; Zhang, X.D.; Yang, L.M.; Wang, F. Effect of structural vacancies on lattice vibration, mechanical, electronic, and thermodynamic properties of Cr5BSi3. Chin. Phys. B 2022, 31, 026101. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Zhang, X.D.; Bi, H.J.; Liu, X.Q.; Wang, F. First-principles prediction of structure, mechanical and thermodynamic properties of BixGeyOz ternary bismuth crystals. Vacuum 2022, 195, 110696. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, H.L.; Gao, S.; Zeng, Q.F. First-principles study of electronic structure and optical properties of Er: Lu2O3. J. Rare Earths 2021, 39, 453–459. [Google Scholar] [CrossRef]
- Bao, L.H.; Ning, J.; Liu, Z.Z. Mechanism for transmittance light tunable property of nanocrystalline Eu-doped SmB6: Experimental and first-principles study. J. Rare Earths 2021, 39, 1100–1107. [Google Scholar] [CrossRef]
- Liu, X.M.; Wang, Q.; Zhao, C.; Li, H.P.; Wang, M.L.; Chen, D.; Wang, H.W. Formation of ordered precipitates in Al-Sc-Er-(Si/Zr) alloy from first-principles study. J. Rare Earths 2021, 39, 609–620. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using vasp: Density-functional theory and beyond. J. Comp. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Akkermans, R.L.; Spenley, N.A.; Robertson, S.H. Monte Carlo methods in materials studio. Mol. Simul. 2013, 39, 1153–1164. [Google Scholar] [CrossRef]
- Hutchinson, J. Singular behaviour at the end of a tensile crack in a hardening material. J. Mech. Phys. Solids 1968, 16, 13–31. [Google Scholar] [CrossRef]
- Rice, J.R.; Rosengren, G. Plane strain deformation near a crack tip in a power-law hardening material. J. Mech. Phys. Solids 1968, 16, 1–12. [Google Scholar] [CrossRef]
- Li, J.; Zhang, M.; Zhou, Y.; Chen, G. First-principles study of Al/A13Ti heterogeneous nucleation interface. Appl. Surf. Sci. 2014, 307, 593–600. [Google Scholar] [CrossRef]
- Siegel, D.J.; Hector, L.G., Jr.; Adams, J.B. Adhesion, stability, and bonding at metal/metal-carbide interfaces: Al/WC. Surf. Sci. 2002, 498, 321–336. [Google Scholar] [CrossRef]
- Rapcewicz, K.; Chen, B.; Yakobson, B.; Bernholc, J. Consistent methodology for calculating surface and interface energies. Phys. Rev. B 1998, 57, 7281. [Google Scholar] [CrossRef]
- Han, Y.; Dai, Y.; Shu, D.; Wang, J.; Sun, B. First-principles study of TiB2 (0001) surfaces. J. Phys. Condens. Matter 2006, 18, 4197. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-S.; Li, L.; Han, J.; Wang, C.-T.; Xiao, Y.-Q.; Jian, X.-D.; Qian, P.; Su, Y.-J. First-Principles study on the nucleation of precipitates in ternary Al alloys doped with Sc, Li, Zr, and Ti elements. Appl. Surf. Sci. 2020, 526, 146455. [Google Scholar] [CrossRef]
- Kaya, M.; Ceyhan, A.A.; Şahin, Ö. Effects of different temperatures and additives on the metastable zone width precipitation kinetics of NaBO2. Russ. J. Phys. Chem. A 2014, 88, 402–408. [Google Scholar] [CrossRef]
- Ozoliņš, V.; Wolverton, C.; Zunger, A. Cu-Au, Ag-Au, Cu-Ag, and Ni-Au intermetallics: First-principles study of temperature-composition phase diagrams and structures. Phys. Rev. B 1998, 57, 6427. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, P.; Lv, H.; Gao, H.; Wang, Y.; Wang, J.; Sun, B. Orientation relationships and interface structure between Al11Ce3 and Al in Al–Ce eutectic. J. Mater. Res. Technol. 2022, 18, 693–704. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Zhou, Z.; Wu, B.; Dou, S.; Zhao, C.; Xiong, Y.; Wu, Y.; Yang, S.; Wei, Z. Thermodynamic Properties of Elements and Compounds in Al-Sc Binary System from Ab Initio Calculations Based on Density Functional Theory. Metall. Mater. Trans. A 2014, 45, 1720–1735. [Google Scholar] [CrossRef]
- Lifshitz, I.M.; Slyozov, V.V. The kinetics of precipitation from supersaturated solid solutions. J. Phys. Chem. Solids 1961, 19, 35–50. [Google Scholar] [CrossRef]
- Wagner, C. Zeitschrift für Elektrochemie. Berichte Bunsenges. Phys. Chem. 1961, 65, 7–8. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Model1 | Model2 | Model3 | Model4 | Model5 | Model6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Al11Sc3 | 0.14 | 5.84 | −0.62 | 5.21 | −0.52 | 5.00 | 0.35 | 12.18 | −0.21 | 6.77 | 0.32 | 11.13 |
| Al11Y3 | 0.42 | 13.56 | 0.05 | 11.97 | 0.11 | 9.62 | 0.79 | 23.08 | −0.03 | 21.30 | 0.83 | 16.95 |
| Al11La3 | 0.56 | 26.09 | 0.43 | 22.30 | 0.52 | 20.79 | 0.98 | 37.28 | 0.21 | 34.12 | 1.04 | 27.30 |
| Al11Ce3 | 0.58; 0.698 [87] | 23.44 | 0.39 | 20.28 | 0.46 | 19.70 | 0.95 | 33.65 | 0.18 | 32.45 | 1.00 | 25.71 |
| Al11Pr3 | 0.57 | 21.67 | 0.38 | 18.04 | 0.43 | 17.86 | 0.95 | 31.09 | 0.15 | 30.23 | 0.99 | 23.58 |
| Al11Nd3 | 0.56 | 19.68 | 0.36 | 16.39 | 0.40 | 15.98 | 0.94 | 29.18 | 0.12 | 28.33 | 0.98 | 21.93 |
| Al11Pm3 | 0.54 | 18.17 | 0.31 | 15.34 | 0.36 | 14.27 | 0.93 | 27.32 | 0.09 | 26.61 | 0.96 | 20.46 |
| Al11Sm3 | 0.52 | 17.14 | 0.28 | 14.11 | 0.31 | 13.01 | 0.91 | 26.10 | 0.07 | 25.39 | 0.94 | 19.42 |
| Al11Eu3 | 0.50 | 15.74 | 0.23 | 12.91 | 0.26 | 11.67 | 0.89 | 24.72 | 0.05 | 23.96 | 0.93 | 18.28 |
| Al11Gd3 | 0.48 | 14.53 | 0.15 | 12.75 | 0.20 | 10.58 | 0.87 | 23.57 | 0.02 | 22.59 | 0.90 | 17.25 |
| Al11Tb3 | 0.46 | 13.65 | 0.09 | 11.80 | 0.16 | 9.74 | 0.84 | 22.78 | −0.01 | 21.57 | 0.87 | 16.70 |
| Al11Dy3 | 0.44 | 12.91 | 0.04 | 11.26 | 0.11 | 9.01 | 0.80 | 22.17 | −0.03 | 20.54 | 0.83 | 16.85 |
| Al11Ho3 | 0.38 | 13.24 | −0.01 | 10.60 | 0.08 | 8.28 | 0.77 | 21.70 | −0.05 | 19.60 | 0.79 | 17.01 |
| Al11Er3 | 0.36 | 12.79 | −0.07 | 9.62 | 0.03 | 7.66 | 0.74 | 21.17 | −0.08 | 18.70 | 0.76 | 16.63 |
| Al11Tm3 | 0.34 | 11.93 | −0.11 | 8.97 | 0.00 | 6.97 | 0.72 | 20.14 | −0.11 | 17.69 | 0.73 | 16.01 |
| Al11Yb3 | 0.32 | 11.36 | −0.15 | 8.43 | −0.01 | 6.53 | 0.70 | 19.12 | −0.12 | 16.31 | 0.71 | 15.37 |
| Al11Lu3 | 0.32 | 10.31 | −0.16 | 7.59 | −0.04 | 5.92 | 0.70 | 17.83 | −0.12 | 14.73 | 0.70 | 14.54 |
| Temperature | 0 K | 100 K | 200 K | 300 K | 400 K | 500 K | 600 K | 700 K | 800 K | 900 K | 1000 K | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| System | ||||||||||||
| Al11Sc3 | −0.16 | −1.21 | −5.20 | −10.45 | −16.50 | −23.30 | −30.93 | −39.54 | −49.35 | −60.65 | −73.30 | |
| Al11Y3 | −346.45 | −344.08 | −335.36 | −324.80 | −313.97 | −303.38 | −293.29 | −283.93 | −275.53 | −268.39 | −262.36 | |
| Al11La3 | −702.96 | −697.85 | −679.25 | −656.42 | −632.40 | −608.05 | −583.72 | −559.63 | −535.97 | −512.93 | −490.22 | |
| Al11Ce3 | −667.30 | −662.57 | −645.30 | −624.09 | −601.72 | −578.99 | −556.21 | −533.58 | −511.25 | −489.40 | −467.66 | |
| Al11Pr3 | −610.96 | −606.44 | −589.96 | −569.69 | −548.32 | −526.60 | −504.85 | −483.24 | −461.94 | −441.10 | −420.37 | |
| Al11Nd3 | −561.88 | −557.56 | −541.80 | −522.49 | −502.21 | −481.71 | −461.31 | −441.20 | −421.58 | −402.66 | −384.14 | |
| Al11Pm3 | −514.29 | −510.20 | −495.30 | −477.09 | −458.04 | −438.87 | −419.92 | −401.40 | −383.52 | −366.54 | −350.19 | |
| Al11Sm3 | −477.42 | −473.59 | −459.64 | −442.59 | −424.79 | −406.92 | −389.30 | −372.16 | −355.71 | −340.21 | −325.42 | |
| Al11Eu3 | −437.48 | −433.74 | −420.08 | −403.43 | −386.11 | −368.80 | −351.83 | −335.43 | −319.85 | −305.36 | −291.77 | |
| Al11Gd3 | −397.41 | −393.80 | −380.62 | −364.55 | −347.87 | −331.24 | −314.99 | −299.37 | −284.61 | −271.02 | −258.41 | |
| Al11Tb3 | −363.03 | −359.65 | −347.24 | −332.14 | −316.51 | −301.01 | −285.96 | −271.60 | −258.20 | −246.05 | −235.01 | |
| Al11Dy3 | −329.73 | −326.52 | −314.68 | −300.26 | −285.34 | −270.57 | −256.25 | −242.64 | −229.98 | −218.59 | −208.30 | |
| Al11Ho3 | −298.29 | −295.33 | −284.30 | −270.84 | −256.94 | −243.22 | −229.99 | −217.50 | −206.01 | −195.83 | −186.82 | |
| Al11Er3 | −267.61 | −264.95 | −254.81 | −242.36 | −229.45 | −216.66 | −204.28 | −192.53 | −181.65 | −171.92 | −163.16 | |
| Al11Tm3 | −239.40 | −236.98 | −227.60 | −216.04 | −204.07 | −192.27 | −180.90 | −170.19 | −160.39 | −151.78 | −144.19 | |
| Al11Yb3 | −207.63 | −205.27 | −196.03 | −184.62 | −172.81 | −161.16 | −149.96 | −139.43 | −129.81 | −121.40 | −114.02 | |
| Al11Lu3 | −184.02 | −182.39 | −175.31 | −166.42 | −157.29 | −148.42 | −140.07 | −132.47 | −125.86 | −120.55 | −116.36 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, W.; Liu, Y.; Lin, L.; Wang, Y.; Chen, L.; Li, Z.; Peng, H.; Fan, T.; Wu, Y.; Deng, Y.; et al. Systematic First-Principles Investigations of the Nucleation, Growth, and Surface Properties of Al11RE3 Second-Phase Particles in Al-Based Alloys. Coatings 2024, 14, 983. https://doi.org/10.3390/coatings14080983
Yin W, Liu Y, Lin L, Wang Y, Chen L, Li Z, Peng H, Fan T, Wu Y, Deng Y, et al. Systematic First-Principles Investigations of the Nucleation, Growth, and Surface Properties of Al11RE3 Second-Phase Particles in Al-Based Alloys. Coatings. 2024; 14(8):983. https://doi.org/10.3390/coatings14080983
Chicago/Turabian StyleYin, Wei, Yuming Liu, Lan Lin, Yiru Wang, Leyi Chen, Zhaoting Li, Honghu Peng, Touwen Fan, Yuanzhi Wu, Yuanxiang Deng, and et al. 2024. "Systematic First-Principles Investigations of the Nucleation, Growth, and Surface Properties of Al11RE3 Second-Phase Particles in Al-Based Alloys" Coatings 14, no. 8: 983. https://doi.org/10.3390/coatings14080983