
The Synergistic Effect of Cross-Linked and Electrostatic Self-Assembly Si/MXene Composites Anode for Highly Efficient Lithium-Ion Battery

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Modification of Nanopowder Silicon
2.2.1. The Pretreatment of Silicon Nanopowder
2.2.2. The Pretreatment of SiOx/Si Nanopowder
2.3. Synthesis of MXene
2.4. Fabrication of nmSi-NH2/MXene Anode
2.5. Assembly of Lithium-Ion Batteries
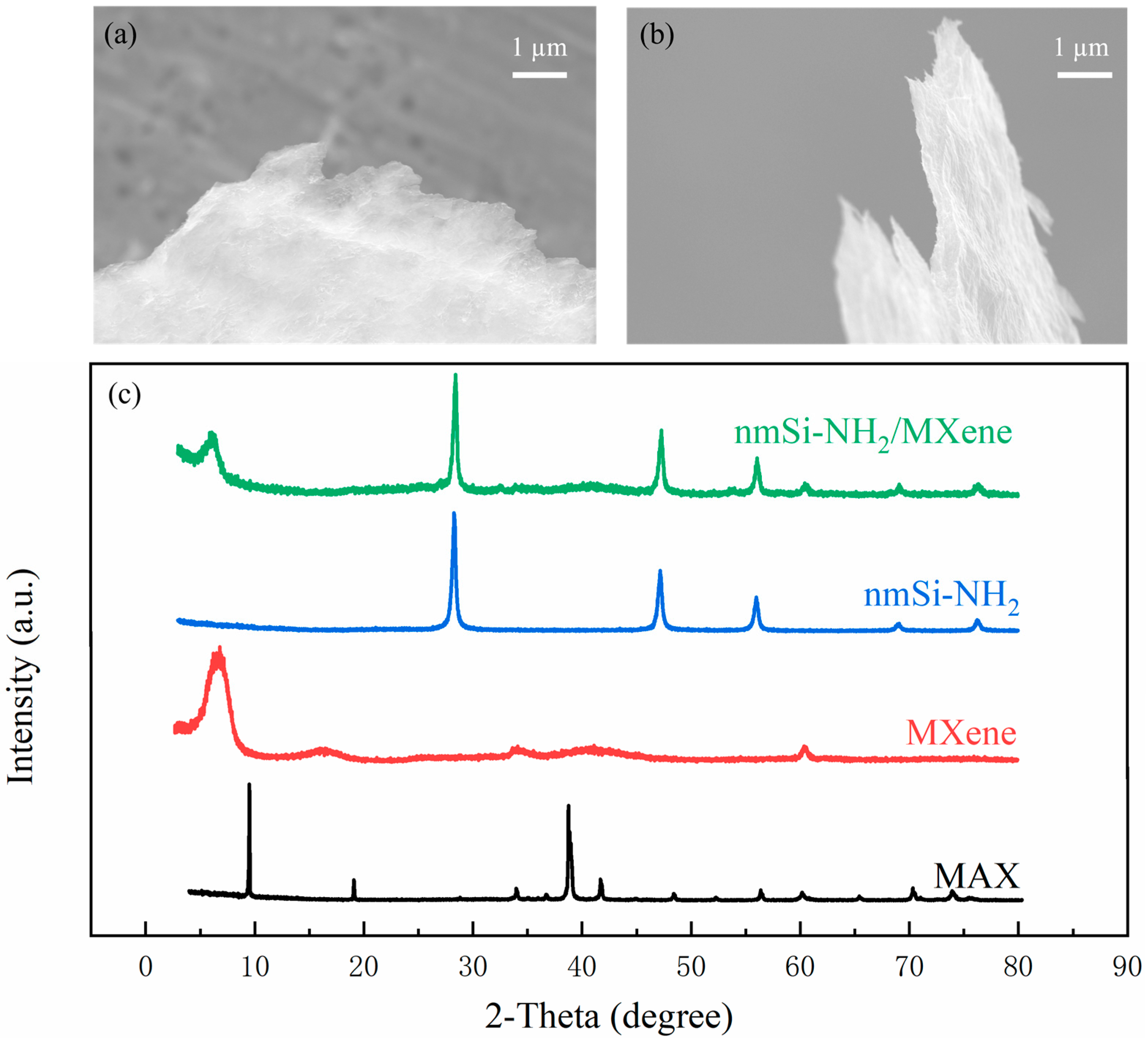
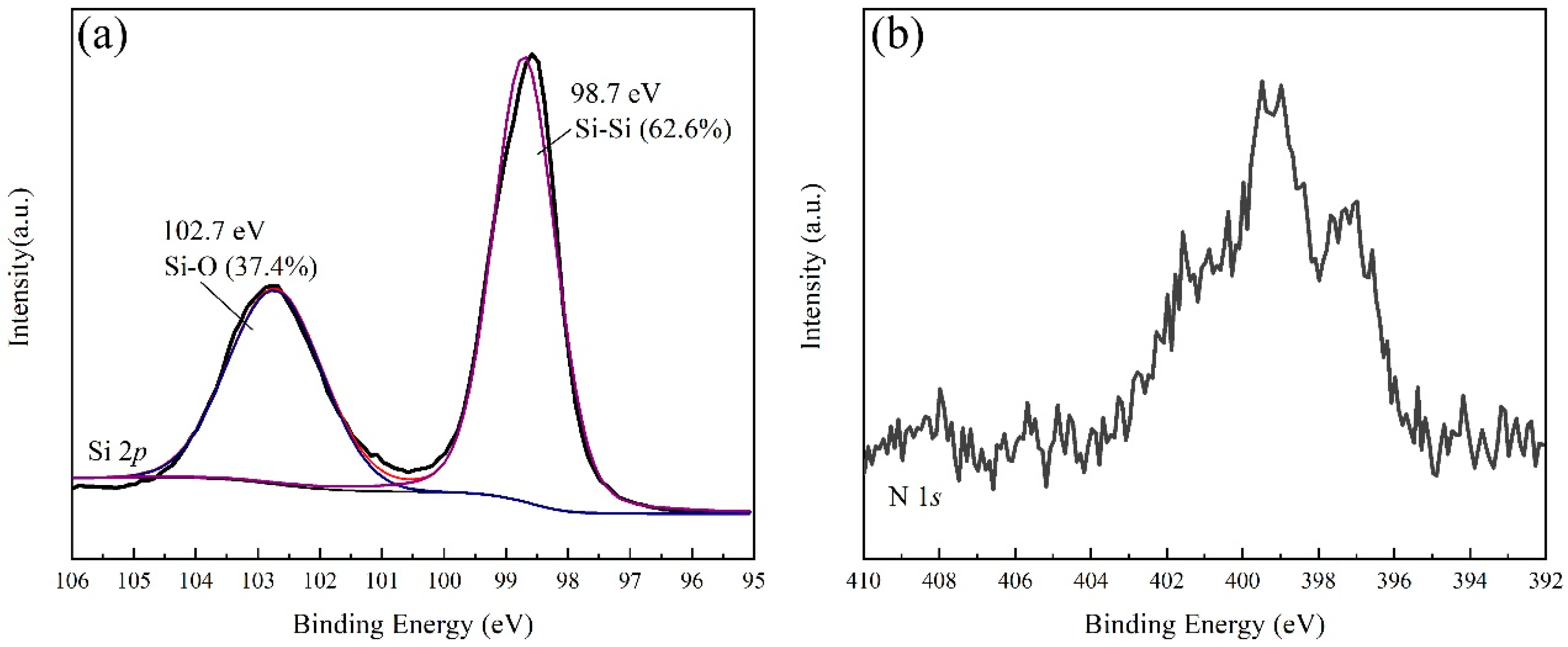
3. Material Characterization
4. Results and Discussion
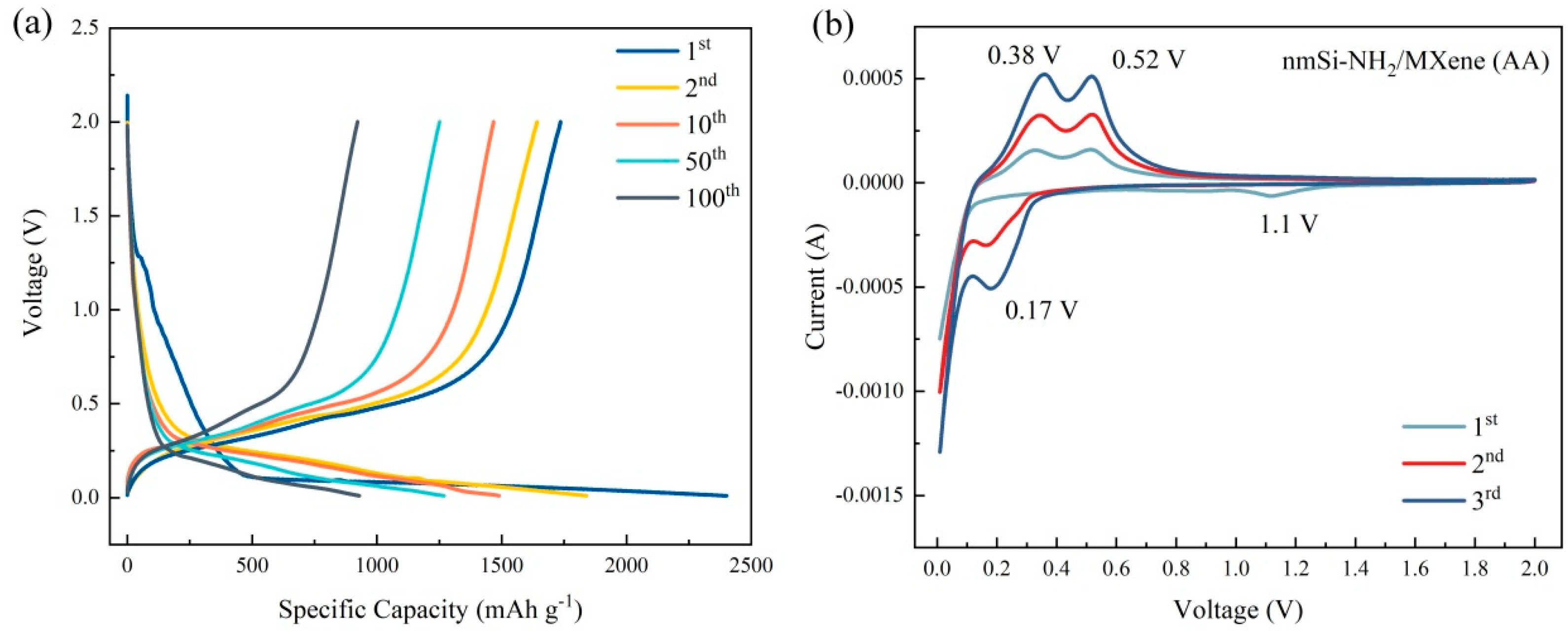
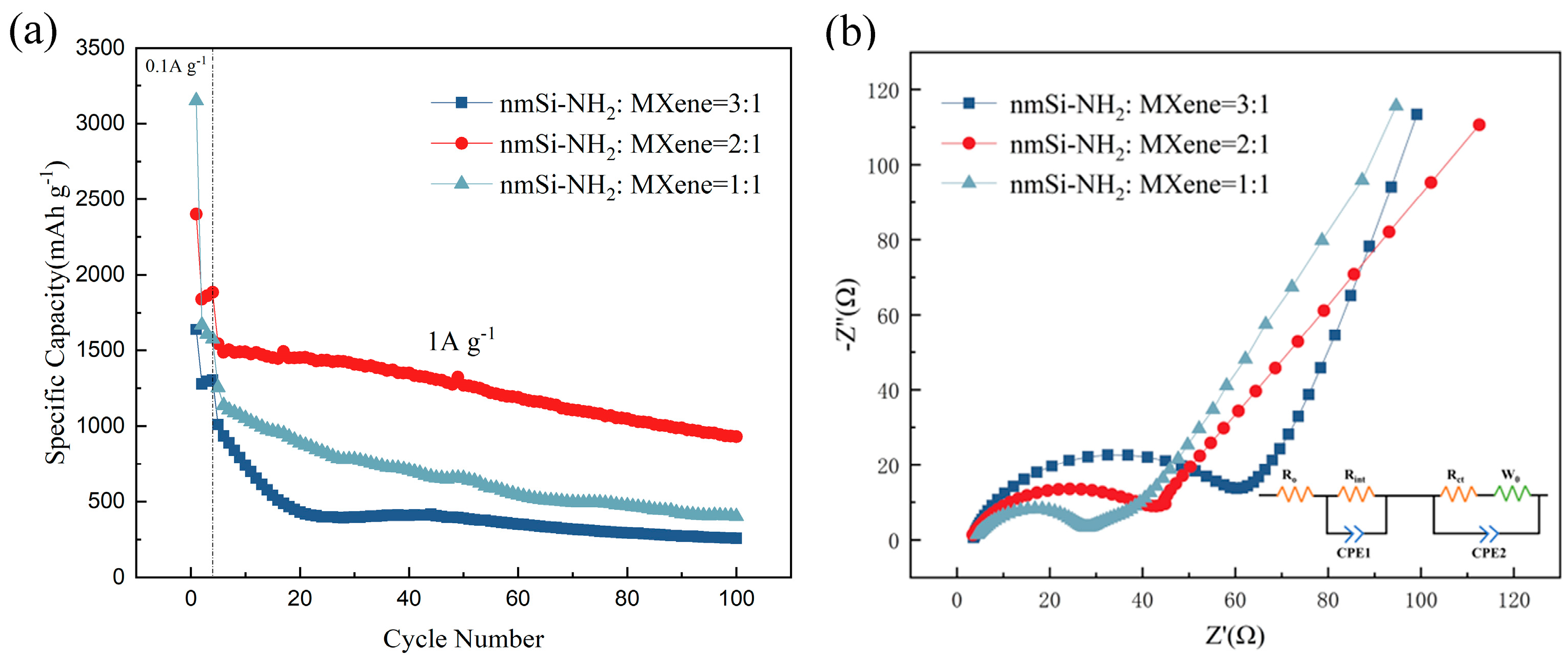
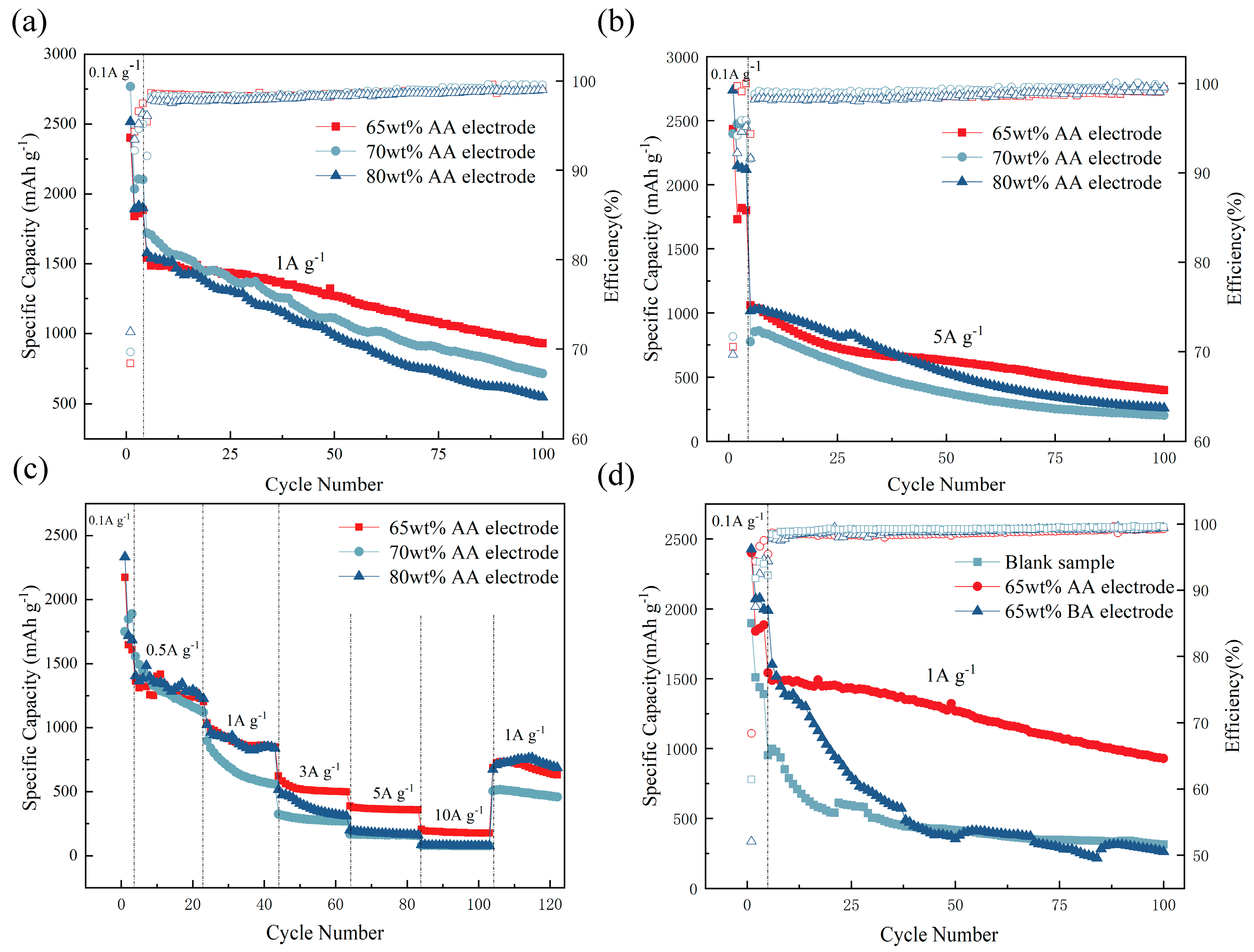
5. Electrochemical Performance
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, M.-S.; Wang, G.-L.; Wang, S.; Zhang, J.; Wang, J.; Zhong, W.; Tang, F.; Yang, Z.-L.; Zheng, J.; Li, X. In Situ Catalytic Growth 3D Multi-Layers Graphene Sheets Coated Nano-Silicon Anode for High Performance Lithium-Ion Batteries. Chem. Eng. J. 2019, 356, 895–903. [Google Scholar] [CrossRef]
- Xing, Y.; Zhang, L.; Mao, S.; Wang, X.; Wenren, H.; Xia, X.; Gu, C.; Tu, J. Core-Shell Structure of Porous Silicon with Nitrogen-Doped Carbon Layer for Lithium-Ion Batteries. Mater. Res. Bull. 2018, 108, 170–175. [Google Scholar] [CrossRef]
- Liu, N.; Lu, Z.; Zhao, J.; McDowell, M.T.; Lee, H.-W.; Zhao, W.; Cui, Y. A Pomegranate-Inspired Nanoscale Design for Large-Volume-Change Lithium Battery Anodes. Nat. Nanotechnol. 2014, 9, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Hong, D.; Choi, S.; Park, S. Synthesis of Ultrathin Si Nanosheets from Natural Clays for Lithium-Ion Battery Anodes. ACS Nano 2016, 10, 2843–2851. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, J.; Mao, S.; Chang, J.; Hallac, P.B.; Fell, C.R.; Metz, B.; Jiang, J.; Hurley, P.T.; Chen, J. Controllable Synthesis of Hollow Si Anode for Long-Cycle-Life Lithium-Ion Batteries. Adv. Mater. 2014, 26, 4326–4332. [Google Scholar] [CrossRef] [PubMed]
- Bogart, T.D.; Oka, D.; Lu, X.; Gu, M.; Wang, C.; Korgel, B.A. Lithium Ion Battery Peformance of Silicon Nanowires with Carbon Skin. ACS Nano 2014, 8, 915–922. [Google Scholar] [CrossRef]
- Chen, S.; Chen, Z.; Xu, X.; Cao, C.; Xia, M.; Luo, Y. Scalable 2D Mesoporous Silicon Nanosheets for High-Performance Lithium-Ion Battery Anode. Small 2018, 14, 1703361. [Google Scholar] [CrossRef]
- Yao, Y.; McDowell, M.T.; Ryu, I.; Wu, H.; Liu, N.; Hu, L.; Nix, W.D.; Cui, Y. Interconnected Silicon Hollow Nanospheres for Lithium-Ion Battery Anodes with Long Cycle Life. Nano Lett. 2011, 11, 2949–2954. [Google Scholar] [CrossRef]
- Park, G.D.; Cho, J.S.; Kang, Y.C. Novel Cobalt Oxide-Nanobubble-Decorated Reduced Graphene Oxide Sphere with Superior Electrochemical Properties Prepared by Nanoscale Kirkendall Diffusion Process. Nano Energy 2015, 17, 17–26. [Google Scholar] [CrossRef]
- Lee, J.K.; Smith, K.B.; Hayner, C.M.; Kung, H.H. Silicon Nanoparticles–Graphene Paper Composites for Li Ion Battery Anodes. Chem. Commun. 2010, 46, 2025–2027. [Google Scholar] [CrossRef]
- Tokur, M.; Algul, H.; Ozcan, S.; Cetinkaya, T.; Uysal, M.; Akbulut, H. Closing to Scaling-Up High Reversible Si/RGO Nanocomposite Anodes for Lithium Ion Batteries. Electrochim. Acta 2016, 216, 312–319. [Google Scholar] [CrossRef]
- Wang, M.-S.; Wang, Z.-Q.; Jia, R.; Yang, Y.; Zhu, F.-Y.; Yang, Z.-L.; Huang, Y.; Li, X.; Xu, W. Facile Electrostatic Self-Assembly of Silicon/Reduced Graphene Oxide Porous Composite by Silica Assist as High Performance Anode for Li-Ion Battery. Appl. Surf. Sci. 2018, 456, 379–389. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, J.; Wang, X.; Qin, J.; Cao, M. A Hybrid Assembly of MXene with NH2−Si Nanoparticles Boosting Lithium Storage Performance. Chem. Asian J. 2020, 15, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jia, Z.; Wang, C.; Feng, A.; Wang, K.; Hou, T.; Liu, J.; Zhang, Y.; Wu, G. Sandwich-like Silicon/Ti3C2Tx MXene Composite by Electrostatic Self-Assembly for High Performance Lithium Ion Battery. Energy 2020, 195, 117047. [Google Scholar] [CrossRef]
- Jin, B.; Wang, D.; Song, L.; Cai, Y.; Ali, A.; Hou, Y.; Chen, J.; Zhang, Q.; Zhan, X. Biomass-Derived Fluorinated Corn Starch Emulsion as Binder for Silicon and Silicon Oxide Based Anodes in Lithium-Ion Batteries. Electrochim. Acta 2021, 365, 137359. [Google Scholar] [CrossRef]
- Su, T.-T.; Ren, W.-F.; Wang, K.; Yuan, J.-M.; Shao, C.-Y.; Ma, J.-L.; Chen, X.-H.; Xiao, L.-P.; Sun, R.-C. Bifunctional Hydrogen-Bonding Cross-Linked Polymeric Binders for Silicon Anodes of Lithium-Ion Batteries. Electrochim. Acta 2022, 402, 139552. [Google Scholar] [CrossRef]
- Tian, Y.; An, Y.; Feng, J. Flexible and Freestanding Silicon/MXene Composite Papers for High-Performance Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2019, 11, 10004–10011. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Zhang, P.; Sun, N.; Anasori, B.; Zhu, Q.-Z.; Liu, H.; Gogotsi, Y.; Xu, B. Self-Assembly of Transition Metal Oxide Nanostructures on MXene Nanosheets for Fast and Stable Lithium Storage. Adv. Mater. 2018, 30, 1707334. [Google Scholar] [CrossRef]
- Ahmed, B.; Anjum, D.H.; Gogotsi, Y.; Alshareef, H.N. Atomic Layer Deposition of SnO2 on MXene for Li-Ion Battery Anodes. Nano Energy 2017, 34, 249–256. [Google Scholar] [CrossRef]
- Ding, Y.; Sun, W.; Yang, W.; Li, Q. Formic Acid as the In-Situ Hydrogen Source for Catalytic Reduction of Nitrate in Water by PdAg Alloy Nanoparticles Supported on Amine-Functionalized SiO2. Appl. Catal. B Environ. 2017, 203, 372–380. [Google Scholar] [CrossRef]
- Bao, S.; Li, J.; Guan, B.; Jia, M.; Terasaki, O.; Yu, J. A Green Selective Water-Etching Approach to MOF@Mesoporous SiO2 Yolk-Shell Nanoreactors with Enhanced Catalytic Stabilities. Matter 2020, 3, 498–508. [Google Scholar] [CrossRef]
- Alkarmo, W.; Aqil, A.; Ouhib, F.; Thomassin, J.-M.; Mazouzi, D.; Guyomard, D.; Detrembleur, C.; Jérôme, C. Nanostructured 3D Porous Hybrid Network of N-Doped Carbon, Graphene and Si Nanoparticles as an Anode Material for Li-Ion Batteries. New J. Chem. 2017, 41, 10555–10560. [Google Scholar] [CrossRef]
- Jin, Y.G.; Qiao, S.Z.; da Costa, J.C.D.; Wood, B.J.; Ladewig, B.P.; Lu, G.Q. Hydrolytically Stable Phosphorylated Hybrid Silicas for Proton Conduction. Adv. Funct. Mater. 2007, 17, 3304–3311. [Google Scholar] [CrossRef]
- Maleski, K.; Mochalin, V.N.; Gogotsi, Y. Dispersions of Two-Dimensional Titanium Carbide MXene in Organic Solvents. Chem. Mater. 2017, 29, 1632–1640. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, Y.; Xu, J. Synthesis of Ag–Ho, Ag–Sm, Ag–Zn, Ag–Cu, Ag–Cs, Ag–Zr, Ag–Er, Ag–Y and Ag–Co Metal Organic Nanoparticles for UV-Vis-NIR Wide-Range Bio-Tissue Imaging. Photochem. Photobiol. Sci. 2019, 18, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Bashouti, M.Y.; Paska, Y.; Puniredd, S.R.; Stelzner, T.; Christiansen, S.; Haick, H. Silicon Nanowires Terminated with Methyl Functionalities Exhibit Stronger Si–C Bonds than Equivalent 2D Surfaces. Phys. Chem. Chem. Phys. 2009, 11, 3845–3848. [Google Scholar] [CrossRef]
- Ahmed, B.; Anjum, D.H.; Hedhili, M.N.; Gogotsi, Y.; Alshareef, H.N. H2O2 Assisted Room Temperature Oxidation of Ti2C MXene for Li-Ion Battery Anodes. Nanoscale 2016, 8, 7580–7587. [Google Scholar] [CrossRef]
- Meng, R.; Huang, J.; Feng, Y.; Zu, L.; Peng, C.; Zheng, L.; Zheng, L.; Chen, Z.; Liu, G.; Chen, B.; et al. Black Phosphorus Quantum Dot/Ti3C2 MXene Nanosheet Composites for Efficient Electrochemical Lithium/Sodium-Ion Storage. Adv. Energy Mater. 2018, 8, 1801514. [Google Scholar] [CrossRef]
- Shi, P.; Xue, R.; Wei, Y.; Lei, X.; Ai, J.; Wang, T.; Shi, Z.; Wang, X.; Wang, Q.; Mohammed Soliman, F.; et al. Gold Nanoparticles/Tetraaminophenyl Porphyrin Functionalized Multiwalled Carbon Nanotubes Nanocomposites Modified Glassy Carbon Electrode for the Simultaneous Determination of p-Acetaminophen and p-Aminophenol. Arab. J. Chem. 2020, 13, 1040–1051. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Byrne, J.A.; McLaughlin, J.; Ahmed, W. Study of Human Serum Albumin Adsorption and Conformational Change on DLC and Silicon Doped DLC Using XPS and FTIR Spectroscopy. J. Biomater. Nanobiotechnol. 2013, 4, 10. [Google Scholar] [CrossRef]
- Siddique, A.B.; Pramanick, A.K.; Chatterjee, S.; Ray, M. Amorphous Carbon Dots and Their Remarkable Ability to Detect 2,4,6-Trinitrophenol. Sci. Rep. 2018, 8, 9770. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.-H.; Kim, K.-H.; Hong, S.-H. An in Situ Formed Graphene Oxide–Polyacrylic Acid Composite Cage on Silicon Microparticles for Lithium Ion Batteries via an Esterification Reaction. J. Mater. Chem. A 2019, 7, 12763–12772. [Google Scholar] [CrossRef]
- Jung, C.-H.; Kim, K.-H.; Hong, S.-H. Stable Silicon Anode for Lithium-Ion Batteries through Covalent Bond Formation with a Binder via Esterification. ACS Appl. Mater. Interfaces 2019, 11, 26753–26763. [Google Scholar] [CrossRef]
- Xie, Y.; Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y.; Yu, X.; Nam, K.-W.; Yang, X.-Q.; Kolesnikov, A.I.; Kent, P.R.C. Role of Surface Structure on Li-Ion Energy Storage Capacity of Two-Dimensional Transition-Metal Carbides. J. Am. Chem. Soc. 2014, 136, 6385–6394. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.E.; Zhao, M.-Q.; Makaryan, T.; Halim, J.; Boota, M.; Kota, S.; Anasori, B.; Barsoum, M.W.; Gogotsi, Y. Porous Two-Dimensional Transition Metal Carbide (MXene) Flakes for High-Performance Li-Ion Storage. ChemElectroChem 2016, 3, 689–693. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, W.; Zhang, X.; He, J.; Chen, J.; Wang, Y.; Cao, T. Directing Silicon–Graphene Self-Assembly as a Core/Shell Anode for High-Performance Lithium-Ion Batteries. Langmuir 2013, 29, 744–749. [Google Scholar] [CrossRef]
- Park, G.D.; Choi, J.H.; Jung, D.S.; Park, J.-S.; Kang, Y.C. Three-Dimensional Porous Pitch-Derived Carbon Coated Si Nanoparticles-CNT Composite Microsphere with Superior Electrochemical Performance for Lithium Ion Batteries. J. Alloys Compd. 2020, 821, 153224. [Google Scholar] [CrossRef]
- Chen, X.; Li, X.; Ding, F.; Xu, W.; Xiao, J.; Cao, Y.; Meduri, P.; Liu, J.; Graff, G.L.; Zhang, J.-G. Conductive Rigid Skeleton Supported Silicon as High-Performance Li-Ion Battery Anodes. Nano Lett. 2012, 12, 4124–4130. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition of nmSi-NH2/MXene | 1 Composition of Slurry | Annealing Condition | Sample Code |
|---|---|---|---|
| 75 wt.% nmSi-NH2–25 wt.%MXene | 65%: 20%: 15% | 180 °C N2 | nmSi-NH2: MXene = 3:1 |
| 66 wt.% nmSi-NH2–33 wt.%MXene | 65%: 20%: 15% | 180 °C N2 | 2 nmSi-NH2: MXene = 2:1 |
| 50 wt.% nmSi-NH2–50 wt.%MXene | 65%: 20%: 15% | 180 °C N2 | nmSi-NH2: MXene = 1:1 |
| 66 wt.% nmSi-NH2–33 wt.%MXene | 65%: 20%: 15% | 180 °C N2 | 65 wt% AA electrode |
| 66 wt.% nmSi-NH2–33 wt.%MXene | 70%: 20%: 10% | 180 °C N2 | 70 wt% AA electrode |
| 66 wt.% nmSi-NH2–33 wt.%MXene | 80%: 10%: 10% | 180 °C N2 | 80 wt% AA electrode |
| 66 wt.% nmSi-NH2–33 wt.%MXene | 65%: 20%: 15% | None | 65 wt% BA electrode |
| 66 wt.% nmSi–33 wt.%MXene | 65%: 20%: 15% | None | Blank sample |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, S.; Liu, C.; Ren, J.; Wang, T.; Geng, X.; Yuan, Y.; Zhao, C.; Zhao, C.; Yang, L. The Synergistic Effect of Cross-Linked and Electrostatic Self-Assembly Si/MXene Composites Anode for Highly Efficient Lithium-Ion Battery. Coatings 2024, 14, 1210. https://doi.org/10.3390/coatings14091210
Kong S, Liu C, Ren J, Wang T, Geng X, Yuan Y, Zhao C, Zhao C, Yang L. The Synergistic Effect of Cross-Linked and Electrostatic Self-Assembly Si/MXene Composites Anode for Highly Efficient Lithium-Ion Battery. Coatings. 2024; 14(9):1210. https://doi.org/10.3390/coatings14091210
Chicago/Turabian StyleKong, Songjia, Chenguang Liu, Jiawei Ren, Tianchang Wang, Xianwei Geng, Yudan Yuan, Chun Zhao, Cezhou Zhao, and Li Yang. 2024. "The Synergistic Effect of Cross-Linked and Electrostatic Self-Assembly Si/MXene Composites Anode for Highly Efficient Lithium-Ion Battery" Coatings 14, no. 9: 1210. https://doi.org/10.3390/coatings14091210
APA StyleKong, S., Liu, C., Ren, J., Wang, T., Geng, X., Yuan, Y., Zhao, C., Zhao, C., & Yang, L. (2024). The Synergistic Effect of Cross-Linked and Electrostatic Self-Assembly Si/MXene Composites Anode for Highly Efficient Lithium-Ion Battery. Coatings, 14(9), 1210. https://doi.org/10.3390/coatings14091210