1. Introduction
The use of raw materials from renewable resources, such as vegetable oils and natural fibers, for the manufacturing of biocomposites has increased at a tremendous rate. The main goal of such an approach is to reduce the environmental footprint left by petroleum-based thermoplastic resins and inorganic engineered fibers. Moreover, the abundance and low cost of renewable raw materials have also positively contributed for the development of biocomposites [
1,
2,
3,
4,
5]. In particular, the substitution of petroleum-based polyols to natural oil-based polyols to produce polyurethanes (PUs) has gained special attention [
6]. PUs are currently very prominent materials due to their wide range of applications and properties, which depend on their synthesis process [
1,
2,
3,
4]. Additionally, composites made from PUs exhibit good mechanical and thermal properties, thereby increasing their versatility [
5,
7].
Vegetable oils are abundantly available, inexpensive, and highly-functional materials. They are composed of triglycerides containing 14–18 carbon-long fatty acid chains, which may have up to three carbon-carbon double bonds [
1,
3,
4,
5]. The most common unsaturated fatty acids found in vegetable oils are linoleic, and linolenic acids [
1,
3,
4,
5]. The scientific literature reports on the preparation of polymers from soybean, linseed, sunflower, palm, cotton, and castor oils through synthetic pathways that involve reactions, such as epoxidation, transesterification, or acrylation [
1,
5]. The possible reactions of a vegetable oil are tightly related to its functional groups, present in the triglyceride unit, which may include carbon-carbon double bonds, ester groups, allylic carbons, and carbonyl groups [
3,
5]. Additionally, straight chain polymerization, many of these groups can lead to crosslinking of the fatty acids present in the oil [
1].
The unsaturated fatty acid chains can be easily epoxidized [
1,
2,
3,
4] with an organic acid in the presence of hydrogen peroxide [
1,
3,
8]. The ring-opening of oxyrane rings gives rise to polyols [
1,
2,
3,
4,
8]. The polyols, along with isocyanates, are the main “ingredients” for the preparation of PUs via polycondensation [
5,
8]. In addition to the chemical structure of the co-monomers, the reaction stoichiometry and morphological aspects, such as degree of crystalinity, as shown for segmented thermoplastic PU elastomers [
9,
10,
11,
12,
13], also plays an important role in the final properties of PU materials. Indeed, it has been recently shown that the molar ratio [NCO]/[OH] is directly related to the crosslink density of jathropa oil methyl ester-based PUs [
14]. It is therefore hypothesized that thermal and mechanical properties of PUs fabricated from oils with a different fatty acid composition than jathropa oil are also related to the [NCO]/[OH] molar ratio employed in the synthesis, and that differences in crosslink density will translate into differences in thermo-mechanical properties. These relationships are verified herein.
The present contribution is aimed at providing a simple and yet very efficient route for the preparation of bio-based PUs and composites with high content of renewable carbon sources. It describes the synthesis of polyols from macauba oil (MO) and their use in the preparation of bio-based PUs. The bio-based PUs are prepared with different methylene diphenyl diisocyanate (MDI) to polyol molar ratios, or else [NCO]/[OH], in order to establish its effect on the PU’s thermal and mechanical properties. Macauba (
Acrocomia aculeata) is a common palm tree found in tropical regions. Its fruit has a high oil content that serves several purposes, such as animal and human feeding, soap precursors, and coatings. The composition of unsaturated fatty acids in macauba oil (MO) is 40%–58% of oleic acid, 3%–18% of linoleic acid, and 2% of linolenic acid [
15,
16]. Due to its high unsaturated fatty acid content, MO is a suitable starting material for the preparation of bio-based PUs. Its unique fatty acid composition suggests that PUs prepared from MO exhibit distinct properties when compared to systems prepared from other vegetable oils. It, therefore, becomes relevant to investigate and establish the thermo-mechanical properties of MO-based PUs, and their relationship with different [NCO]/[OH] molar ratios. Indeed, it is expected that with a different degree of unsaturation in comparison to other oils, the MO-based PUs prepared in this work exhibit distinct crosslink densities and thermo-mechanical properties than previously observed with other oils. The proposition of MO as a new bio-renewable building block for PUs requires a careful investigation of the relationships between the [NCO]/[OH] molar ratios employed during the synthesis and the final properties obtained for these new materials.
Since only few low-cost applications are currently being sought for this abundant and underused oil, it is believed that the use of MO in polymeric systems hasn’t reached its full potential yet, making it an ideal candidate for being studied as a possible precursor in the preparation of bio-based PUs and composites. Additionally, it is reported the preparation of biocomposites with bio-based PUs reinforced with coconut husk fibers. Coconut husk fibers are extracted from the seed of the coconut palm (
Cocos nucifera), and its predominant composition is 37–44 wt % of cellulose, 33 wt % of lignin, and 12–20 wt % of hemicellulose, which may vary according to the seasonal conditions, age, and variety of the plant [
6,
17,
18,
19]. Coconut husk fibers are inexpensive agricultural and agro-industrial residues and their use in such composites can significantly reduce their environmental impact [
6,
17]. The use of coconut husk fibers as reinforcement in MO-based PU composites constitutes a potential alternative to more costly systems prepared with bio-based PUs recently investigated, such as soy polyol-based PUs [
20] and castor oil hyperbranched PUs reinforced with graphene [
21].
It is important to point out that the fibers were used
in natura, without the addition of any compatibilizer in the composites formulations. Although these strategies could aid in the mixing of components and further improve the matrix-filler interface, they may not play a role in some systems. For example, as reported by Castro
et al. [
22], the use of castor oil as a compatibilizer for the preparation of nanocomposites of green polyethylene and cellulose nano-crystals has no influence over the storage modulus and tan δ. Moreover, as it has been pointed out by Mazzon
et al. [
23] that biomass-derived epoxy foams can be designed with specific properties, such as
Tg, stiffness, density, and morphology, provided that the cure obeys rigorous equilibrium rules. In this sense, the approach reported herein is less restrictive than others but still shows great potential for improvements.
All materials prepared herein are characterized by Fourier-transform infrared spectroscopy (FTIR), thermogravimetry (TG), derivative thermogravimetry (DTG), differential thermal analysis (DTA), dynamic mechanical analysis (DMA), and scanning electron microscopy (SEM). The role of [NCO]/[OH] molar ratio in the thermo-mechanical properties of bio-based PUs and composites is thoroughly investigated. The influence of fiber-matrix interactions in the composites’ end properties is also explored.
3. Results and Discussion
The use of vegetable oils for the synthesis of polymeric materials has significantly increased since 2000. The polymerization of various unsaturated oils can be achieved by taking advantage of carbon-carbon double bonds in the oils’ chemical structure through various processes. With 39% of its fatty acid chains being oleic acid, and 5% being linoleic acid [
27], the chemical structure of MO shows potential for polymerization through the preliminary preparation of polyols and their subsequent polycondensation with diisocyanates for the synthesis of bio-based PUs.
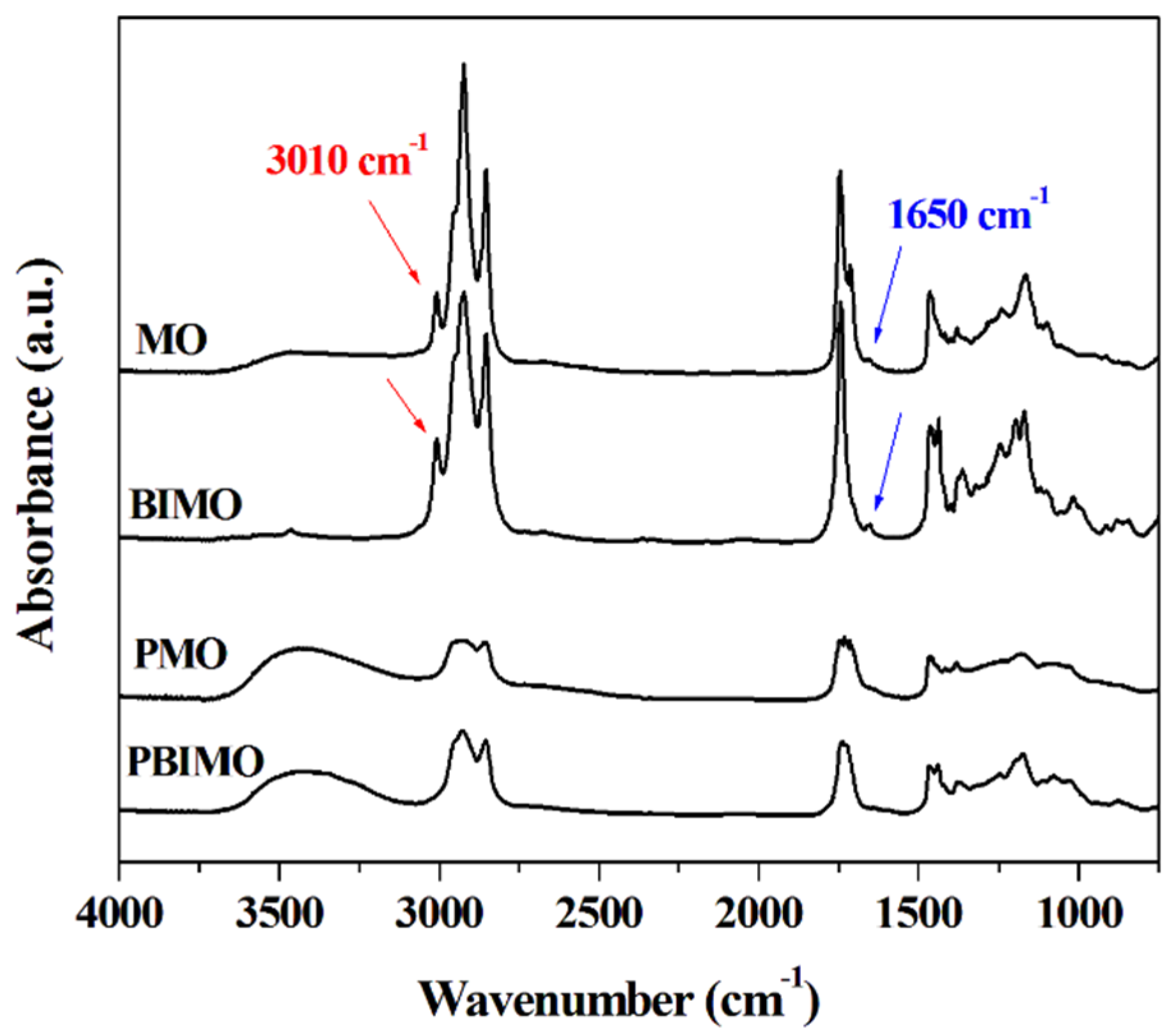
The FTIR spectrum of MO (
Figure 1) exhibits characteristic peaks corresponding to C=C–H vibrations at 1650 cm
−1 and 3010 cm
−1, as well as peaks related to C–H symmetric and asymmetric stretching of aliphatic chains (2924 cm
−1, 2850 cm
−1, respectively), and C=O stretch deformation at approximately 1746 cm
−1 [
28,
29,
30]. On the FTIR spectrum of BIMO, the product of the transesterification of MO with methanol, a change on the profile of the peaks in the 1000–1400 cm
−1 range can be observed, along with the appearance of a typical ester band at 1035 cm
−1 (
Figure 1) [
31,
32]. These differences between the FTIR spectra of MO and BIMO suggest that the methanolysis reaction was successful. The polyol formation for both MO and BIMO (PMO and PBIMO, respectively) is verified by the appearance of a broad band at 3380 cm
−1, characteristic of the O–H stretch of alcohols (
Figure 1), in addition to the disappearance of the peaks at 1650 cm
−1 and 3009 cm
−1, which are related to unsaturations of the fatty acid chains [
3].
Figure 1.
FTIR spectra of macauba oil (MO), polyol from macauba oil (PMO), macauba oil methyl esters (BIMO), and polyol from macauba oil methyl esters (PBIMO).
Figure 1.
FTIR spectra of macauba oil (MO), polyol from macauba oil (PMO), macauba oil methyl esters (BIMO), and polyol from macauba oil methyl esters (PBIMO).
The successful conversion of PMO and PBIMO into PUs with varying [NCO]/[OH] molar ratios was also verified by FTIR spectroscopy (
Figure S1). The FTIR spectra of all PUMO and PUBIMO samples prepared exhibit bands at approximately 3300 cm
−1 and 1530 cm
−1, corresponding to N–H stretching, and deformation of urethane groups, respectively [
33], in addition to bands at 1084–1064 cm
−1, assigned to N–C(O)–O deformations. The band at 3300 cm
−1 related to the N–H stretching can be ascribed to the absorption associated to N–H participating in hydrogen bonding as proton donor, which is expected in the hard segments of polyurethanes [
34]. The band at 2273 cm
−1 is associated with the vibration of free isocyanate groups (–N=C=O). The presence of this band confirmed that not all of the isocyanate groups were involved in the condensation polymerization during the PU cure [
35]. In the 1750–1700 cm
−1 band, there were two overlapping bands; one dominant peak at approximately 1740 cm
−1 corresponding to “free” C=O, and one broad shoulder at approximately 1710 cm
−1, corresponding to a C=O electron donor stretching associated to inter-urethane hydrogen-bonding [
34,
36]. The bands at 900–675 cm
−1 correspond to vibrations of axial deformation characteristic of the angular deformation outside of the plane of the C–H bond of aromatic rings [
35,
37,
38], reminiscent of the MDI structure and clearly shown in the spectrum in
Figure S2.
Figure 2 and
Figure 3 present the FTIR spectra of coconut husk fiber composites prepared from the PUMO and PUBIMO samples. It can be observed that the composites have very similar absorption bands to those of the unreinforced PUs (
Figure S1). The major components of ligno-cellulosic materials are cellulose, hemicellulose, and lignin. The major absorption bands observed in these components are regarded to O–H stretching (3400–3200 cm
−1), C=O stretching (1776–1715 cm
−1), C–O–C (1270 cm
−1) and C–OH (~1050 cm
−1) deformations. The most intense band attributed to O–H and C–O–C is found in cellulose and lignin. Other peaks, due to hydroxyl groups of cellulose, appear at 1360 cm
−1 and 1320 cm
−1 [
39,
40]. It has been proposed that chemical interactions between the fibers and the PUs matrix can potentially occur through bonds between hydroxyl groups of cellulose and carbonyl groups of the PU, and they are responsible for increasing the interfacial adhesion between filler and matrix [
35,
41]. The shift of the band at approximately 3300 cm
−1 for values above 3400 cm
−1 suggests the disruption of a significant amount of hydrogen bonds, resulting in a band for “free” N–H stretching [
34]. However, C=O stretching is still observed on the two overlapping bands, in the 1750–1700 cm
−1 range [
34,
36], which can be explained by the substitution of the urethane NH with OH from cellulose. The remaining isocyanate band at 2273 cm
−1 on the FTIR of the composites (
Figure 2 and
Figure 3) suggests that there is little interaction between “free” isocyanate groups in PU and hydroxyl groups present in the cellulose of coconut husk fibers [
35]. However, the remaining isocyanate band at 2273 cm
−1 on the FTIR of the composites (
Figure 2 and
Figure 3) suggests that there is little interaction between isocyanate groups in PU and hydroxyl groups present in the cellulose of coconut husk fibers [
35]. As will be discussed later in the text, non-favorable reinforcement-matrix interactions have also been detected by SEM. This observation is not completely unexpected since cellulose fibers exhibit an intricate structure with nanofibrils tightly connected through hydrogen bonding. Hence, the availability of hydroxyl groups from the fibers to react with isocyanate groups is very limited.
Figure 4 presents TG and DTG curves for MO, PMO, BIMO and PBIMO samples. The TG curve of MO exhibits two stages of thermal decomposition, with
Tonset at approximately 268 °C and discrete mass loss (17%), indicating the start of oxidation of fatty acids present in the oil. The second degradation step occurs with
Tonset at 396 °C and can be associated with the cleavage of carbon-carbon bonds along the fatty acid chains. These two steps are associated with heat release (Δ
H = −309 J·g
−1), as determined by DTA (
Table 1). PMO also exhibits two stages of thermal decomposition (
Figure 4). The first stage exhibits a small endothermic event near 106 °C (Δ
H = 188 J·g
−1,
Table 1), possibly due to the cleavage of hydrogen bonds present in the polyol, along with the release of water, which explains the lower
Tonset. A pronounced second decomposition step, similar to the second degradation step of MO, can be observed for PMO (
Figure 4 and
Table 1), which suggests decomposition of the functionalized fatty acid chains. The higher thermal stability observed for MO in comparison to PMO can be directly correlated to their chemical structure. As a matter of fact, C=C (614.2 kJ·mol
−1) and C–H (413.4 kJ·mol
−1) bonds predominate in MO, while most of the C=C bonds are replaced with C–OH bonds (353.5 kJ·mol
−1) in PMO, leading to an overall lower thermal stability [
3,
42].
Figure 2.
FTIR spectra of PUMO-11.3, PUMO-11.3-10, and PUMO-11.3-20.
Figure 2.
FTIR spectra of PUMO-11.3, PUMO-11.3-10, and PUMO-11.3-20.
Figure 3.
FTIR spectra of PUBIMO-11.3, PUBIMO-11.3-10, and PUBIMO-11.3-20.
Figure 3.
FTIR spectra of PUBIMO-11.3, PUBIMO-11.3-10, and PUBIMO-11.3-20.
Figure 4.
TG and DTG curves of MO, PMO, BIMO, and PBIMO, acquired at a heating rate of 10 °C·min−1.
Figure 4.
TG and DTG curves of MO, PMO, BIMO, and PBIMO, acquired at a heating rate of 10 °C·min−1.
Table 1.
Tonset, Td, ΔH and mass loss of MO, PMO, BIMO and PBIMO.
Table 1.
Tonset, Td, ΔH and mass loss of MO, PMO, BIMO and PBIMO.
| Material | Tonset (°C) | Td (°C) a | ΔH (J·g−1) | Mass Loss (%) |
|---|
| MO | 268 | 289 | −309 | 17 |
| 396 | 408 | – | 84 |
| PMO | 106 | 112 | 188; −294 | 8 |
| 368 | 401 | – | 83 |
| BIMO | 233 | 253 | 359;−791 | 98 |
| PBIMO | 288 | 315 | 423; −412 | 99 |
BIMO and PBIMO started their thermal decomposition at 233 °C and 288 °C, with temperatures of maximum degradation rate (
Tds) at 253 °C and 315 °C, respectively (
Figure 4 and
Table 1). The single degradation stage is associated with decomposition of the methyl esters with formiate/hydroxyl and epoxy groups. In this decomposition, an endothermic process (∆
H = 359 J·g
−1) occurs followed by an exothermic (∆
H = −791 J·g
−1) (
Table 1). PBIMO is more thermally stable than BIMO according to the data on
Table 1, suggesting that the thermal degradation mechanisms of these materials are distinct. The increase in thermal stability of PBIMO is confirmed by the greater amount of heat involved in its endothermic process (∆
H = 423 J·g
−1) when compared to BIMO (∆
H = 359 J·g
−1). However, in the exothermic event of PBIMO, the heat release (∆
H = −412 J·g
−1) is lower than for BIMO (∆
H = −791 J·g
−1,
Table 1). BIMO is less thermally stable than MO due to its lower molecular weight. However, the epoxidation/hydroxylation reaction of BIMO to produce PBIMO results in an increased thermal stability due to the formation of hydrogen bonds in the polyol. The ∆
H values corresponding to the decomposition processes of PUs and composites (not shown) are not very defined, and therefore don’t provide relevant information about the materials prepared.
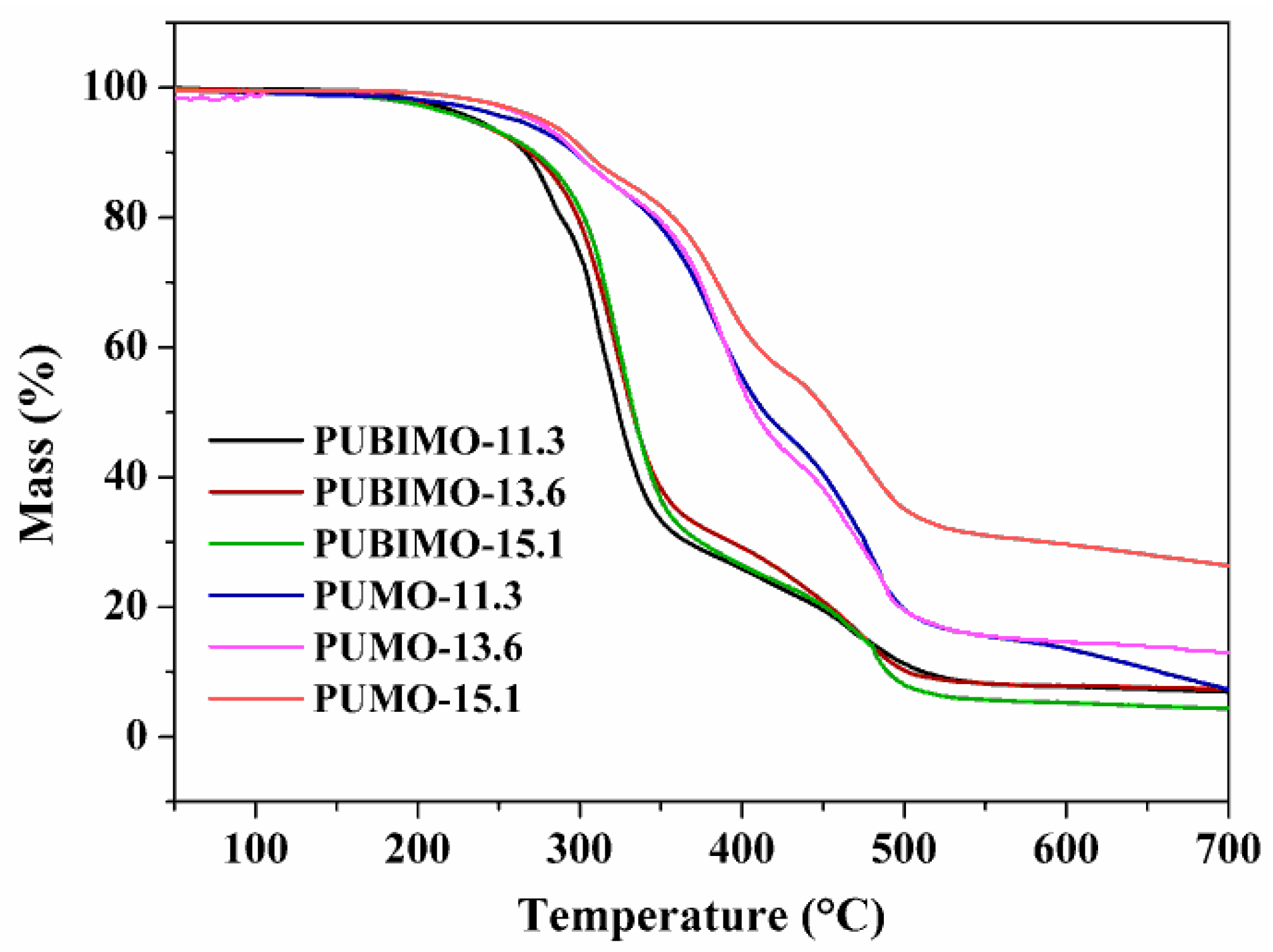
Figure 5 shows the TG curves for PUMO and PUBIMO in [NCO/OH] molar ratios of 11.3, 13.6 and 15.1. The profile of these curves suggests that the thermal decomposition occurs in two stages for PUBIMO-13.6 and PUBIMO-15.1, and three stages for PUBIMO-11.3 and all PUMO samples. The first stage of thermal decomposition of PUBIMO-11.3 and all PUMO samples is discrete in comparison to the other two stages. Therefore, the two subsequent stages are considered as the main degradation steps of the PUs prepared in this work. The first main degradation step, related to the thermal decomposition of urethane bonds, was more pronounced than the second main degradation step, related to the formation of primary and secondary amines. According to the data presented in
Table 2, PUBIMO-13.6 and PUBIMO-15.1 are more thermally stable than PUMO in the same molar ratios. Overall, no coherent trends in the thermal stability of PUs are observed with variations in the [NCO]/[OH] molar ratios.
Figure 6a displays TG and DTG curves of PUMO-11.3 and its composites with 10 wt % and 20 wt % coconut husk fibers (PUMO-11.3-10 and PUMO-11.3-20, respectively).
Table 2 collects the respective degradation temperatures and mass losses. Both data reveal that PUMO-11.3-20 is thermally more stable than PUMO-11.3-10. Indeed, an increase in thermal stability is expected upon reinforcement of PUs with coconut husk fibers, especially in the first decomposition stage. The decomposition of the composites occurs in two stages. The first stage, with
Tds 363 °C and 393 °C for PUMO-11.3-10 and PUMO-11.3-20, respectively, corresponds to the decomposition of urethane groups in the PU, and cellulose and hemicellulose from the fibers [
35]. The second decomposition step, with
Td ~ 460 °C for the two composites, corresponds to the structural degradation of the fatty acid chains and lignin [
43].
Figure 5.
TG curves of PUBIMO-11.3, PUBIMO-13.6, PUBIMO-15.1, PUMO-11.3, PUMO-13.6, and PUMO-15.1, acquired at a heating rate of 10 °C·min−1.
Figure 5.
TG curves of PUBIMO-11.3, PUBIMO-13.6, PUBIMO-15.1, PUMO-11.3, PUMO-13.6, and PUMO-15.1, acquired at a heating rate of 10 °C·min−1.
Table 2.
Tonset, Td, and mass loss of PUMO and PUBIMO in [NCO/OH] molar ratios of 11.3, 13.6, and 15.1; PUMO-11.3 and PUBIMO-11.3 with 10 wt % and 20 wt % of coconut husk fibers.
Table 2.
Tonset, Td, and mass loss of PUMO and PUBIMO in [NCO/OH] molar ratios of 11.3, 13.6, and 15.1; PUMO-11.3 and PUBIMO-11.3 with 10 wt % and 20 wt % of coconut husk fibers.
| Material | Tonset (°C) | Td (°C) a | Mass Loss (%) |
|---|
| PUMO-11.3 | 322 | 381 | 50 |
| 469 | 484 | 33 |
| PUMO-11.3-10 | 311 | 363 | 40 |
| 449 | 462 | 58 |
| PUMO-11.3-20 | 324 | 393 | 53 |
| 444 | 459 | 35 |
| PUMO-13.6 | 277 | 299 | 17 |
| 363 | 390 | 74 |
| PUMO-15.1 | 279 | 303 | 13 |
| 363 | 384 | 27 |
| 496 | 477 | 30 |
| PUBIMO-11.3 | 269 | 303 | 61 |
| 536 | 603 | 29 |
| PUBIMO-11.3-10 | 323 | 328 | 67 |
| 464 | 469 | 21 |
| PUBIMO-11.3-20 | 280 | 327 | 59 |
| 408 | 469 | 19 |
| PUBIMO-13.6 | 296 | 319 | 68 |
| 440 | 478 | 23 |
| PUBIMO-15.1 | 308 | 326 | 71 |
| 476 | 481 | 21 |
Figure 6.
TG and DTG curves of: (a) PUMO-11.3, PUMO-11.3-10, and PUMO-11.3-20; (b) PUBIMO-11.3, PUBIMO-11.3-10, and PUBIMO-11.3-20, acquired at a heating rate of 10 °C·min−1.
Figure 6.
TG and DTG curves of: (a) PUMO-11.3, PUMO-11.3-10, and PUMO-11.3-20; (b) PUBIMO-11.3, PUBIMO-11.3-10, and PUBIMO-11.3-20, acquired at a heating rate of 10 °C·min−1.
A comparison of the TG and DTG curves of PUBIMO-11.3, PUBIMO-11.3-10, and PUBIMO-11.3-20 are presented in
Figure 6b. While three stages of thermal decomposition are observed for PUBIMO-11.3 and PUBIMO-11.3-20, only two degradation steps are observed for PUBIMO-11.3-10. Both composites exhibit
Td ~ 328 °C for the first stage and
Td ~ 469 °C for the second stage. Furthermore, a comparison of the DTG curves of the composites reveals that
Tonset (
Table 2) at each peak does not change significantly, indicating that the addition of fibers did not alter the thermal stability of the materials on a measurable level, possibly because of limited interactions between the polymer matrix and the coconut husk fibers.
Mechanical properties of PUs from MO, and their corresponding composites, were obtained through DMA. The results are shown in
Table 3. It is worth noting that even though PUs and composites from BIMO have been successfully prepared, they consistently broke during the initial cooling of the DMA testing, making the acquisition of mechanical data for these samples impossible.
Table 3.
DMA results for PUs from MO and their corresponding composites.
Table 3.
DMA results for PUs from MO and their corresponding composites.
| Entry | Sample | Tg (°C) | E′ at 25 °C (MPa) | Extractables (wt%) a |
|---|
| 1 | PUMO-11.3 | 68 | 20.1 | 21 |
| 2 | PUMO-13.6 | 76 | 12.2 | 16 |
| 3 | PUMO-15.1 | 106 | 16.1 | 11 |
| 4 | PUMO-11.3-10 | −14; 55 | 33.4 | – |
| 5 | PUMO-11.3-20 | −2; 53 | 24.8 | – |
The role played by the [NCO]/[OH] molar ratio on the
Tg of PUs can be clearly seen in
Table 3 (entries 1–3). As mentioned previously, an increase in the crosslink density of the PU is expected with an increase in the [NCO]/[OH] molar ratio. The increase in crosslink density with [NCO]/[OH] molar ratio can be indirectly estimated from the percentage of extractable materials (non-crosslinked) obtained after Soxhlet extraction of the PUs with methylene chloride for 24 h (
Table 3, entries 1–3). It can be clearly seen in
Table 3 that the percentage of extractable materials decreases as the [NCO]/[OH] molar ratio increases. Such an increase in crosslink density also translates into an increase of the
Tg. Nevertheless, the increase in
Tg is not accompanied by a consistent change in the storage modulus (
Eʹ) at 25 °C. A significant excess of [NCO] with respect to [OH] had to be employed during PU synthesis due to the low functionality of MO, and imposes a limiting factor for [NCO]/[OH] molar ratio variations from a practical standpoint. Due to the fact that the [NCO]/[OH] molar ratio variations herein are not strikingly large, and due to unavoidable sample heterogeneity, it is possible that the proposed trend in crosslink density is not reflected in
E′. As a matter of fact, although significant, the change in
E′ with [NCO]/[OH] molar ratio is fairly small, compared to the improvement seen when PUMO-13.1 is reinforced with coconut husk fibers (entries 1, 4, and 5,
Table 3).
Another interesting observation from the reinforcement of PUs with coconut husk fibers is the appearance of two
Tgs, which indicates a clear interference of the fibers with the condensation polymerization reaction. Similar effects have also been observed during the free radical polymerization of conjugated linseed oil in the presence of rice hulls, and have been attributed to the dispersion of co-monomers during the cure of the composite [
44]. Similar to the change in [NCO]/[OH] molar ratios, an increase in filler load from 10 wt % to 20 wt % does not result in the expected increase in storage modulus. An uneven dispersion of the fibers throughout the composites may have led to such results. The dispersion of the fibers will be discussed later in the text. The foam aspect of the PUs and the size of the molds used made it impossible to conduct tensile tests. However, one can appreciate the reinforcing effect of coconut husk fibers when comparing the storage modulus of PUMO-11.3, PUMO-11.3-10, and PUMO-11.3-20 (
Table 3, entries 1, 4, and 5), as mentioned previously. Indeed, a minimum improvement of approximately 23% in
E′ at 25 °C is obtained when incorporating coconut husk fibers in the PU matrix.
The effect of adding coconut husk fibers to PUMO-13.1 can be better visualized in
Figure 7, which presents a comparison of the DMA results for the PU with and without the reinforcement. In addition to the already mentioned increase in
Eʹ at 25 °C and the appearance of two
Tgs with the addition of the fibers, it can be seen that the unreinforced sample broke prematurely (~120 °C,
Figure 7), whereas the composite endured the full temperature range tested (−60–200 °C). Such behavior shows that the fibers have a positive impact on the ability of the material in supporting the repetitive stress imposed during the DMA test.
Figure 7 also shows an inversion in the expected trend for
E′ between 40 and 70 °C, which reflects the lower
Tgs observed for the composite in comparison to the unreinforced PU. As discussed previously, it is likely that the presence of the fibers during curing of the PU interferes with the condensation polymerization of the resin components, resulting in the unexpected trend in
Eʹ and
Tg.
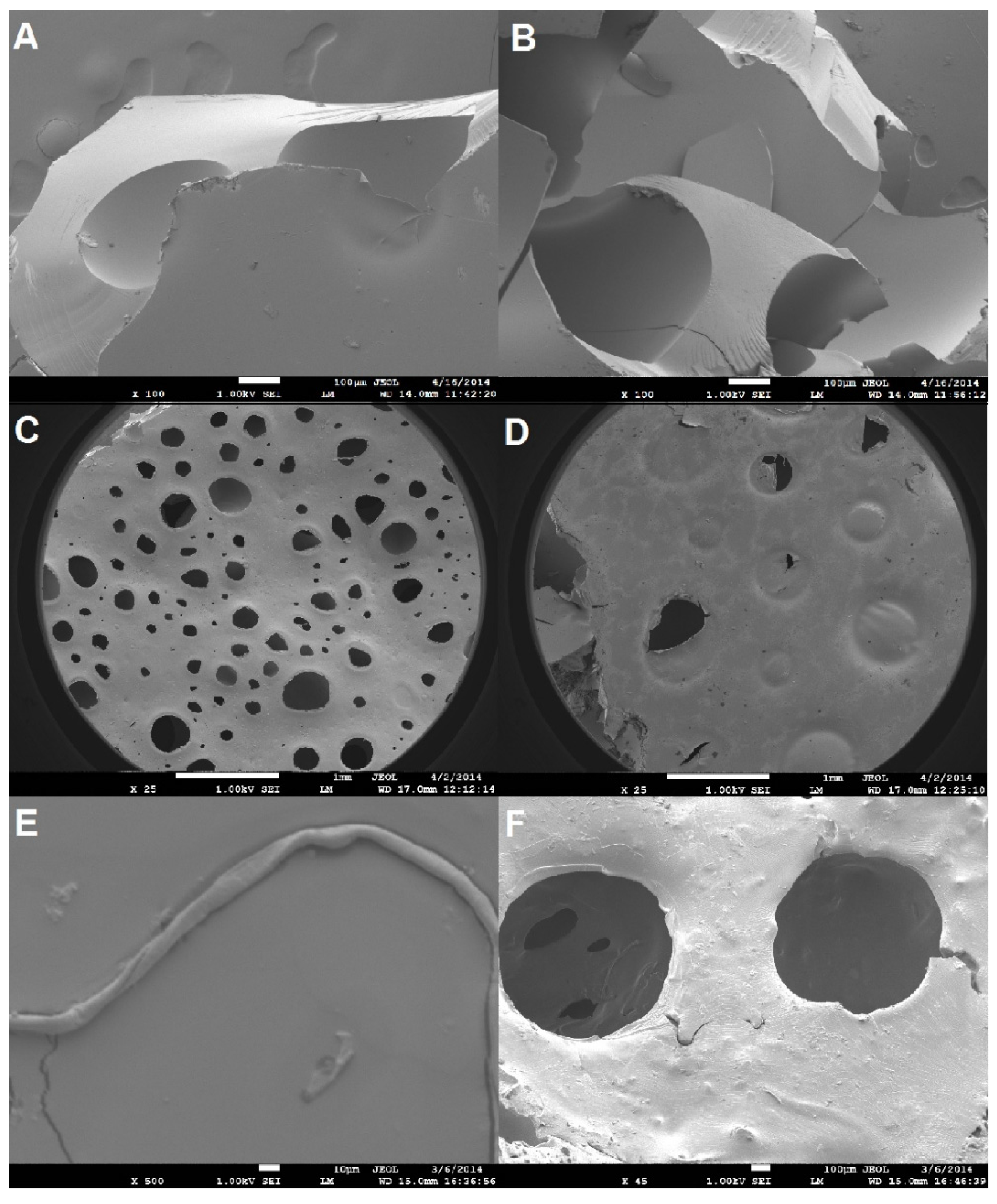
SEM was used to assess morphological features of different PUs and composites.
Figure 8 presents a visual comparison of select images obtained in this study. Overall, the images show poor fiber-matrix interaction, varying pore size distribution, and fracture features of flexible materials.
Figure 7.
DMA results for PUMO-13.1 e PUMO-13.1-10.
Figure 7.
DMA results for PUMO-13.1 e PUMO-13.1-10.
Figure 8.
SEM images of (A) cryo-fracture surface of PUBIMO-15.1 at 100× (the scale bar corresponds to 100 µm); (B) cryo-fracture surface of PUBIMO-13.6 at 100× (the scale bar corresponds to 100 µm); (C) PUMO-11.3 at 25× (the scale bar corresponds to 1 mm); (D) PUBIMO-15.1 at 25× (the scale bar corresponds to 1 mm); (E) PUBIMO-11.3-20 at 500× (the scale bar corresponds to 10 µm); and (F) PUBIMO-11.3-20 at 45× (the scale bar corresponds to 100 µm).
Figure 8.
SEM images of (A) cryo-fracture surface of PUBIMO-15.1 at 100× (the scale bar corresponds to 100 µm); (B) cryo-fracture surface of PUBIMO-13.6 at 100× (the scale bar corresponds to 100 µm); (C) PUMO-11.3 at 25× (the scale bar corresponds to 1 mm); (D) PUBIMO-15.1 at 25× (the scale bar corresponds to 1 mm); (E) PUBIMO-11.3-20 at 500× (the scale bar corresponds to 10 µm); and (F) PUBIMO-11.3-20 at 45× (the scale bar corresponds to 100 µm).
Figure 8A,B shows the top fractured edge view of PUBIMO-15.1 and PUBIMO-13.6, respectively. A comparison of the fractured surfaces in both samples indicates minimum morphological difference when the [NCO]/[OH] molar ratio is varied. The crease lines observed on either image portray the semi-flexible nature of the PUs made from BIMO polyols. As a result of the residual water detected in the polyol from MO, as described when discussing the TG results, PUs made from triglycerides exhibit a significantly increased number of cells when compared to PUs made from BIMO polyols. This trend can be clearly seen in
Figure 8C,D, which depicts PUMO-11.3 and PUBIMO-15.1, respectively.
Figure 8E displays the poor interaction between the coconut fiber and the matrix in PUBIMO-11.3-20. No PU artifacts are found on the fibers identified in the composite samples analyzed. Despite the potential condensation between isocyanate groups and the ligno-cellulosic fibers during the synthesis of the composites, it is hypothesized that the PU network formed is highly hydrophobic due to the long fatty acid chains and low functionality in MO, contrasting with the hydrophilic nature of the coconut husk fibers used as reinforcement. In
Figure 8F, a satisfactory horizontal fiber distribution can be observed throughout the composite with only a few visible fibers sparsely distributed. The low number of fibers identified suggests an uneven vertical fiber distribution. Indeed, settling of the fibers may have occurred during the final cure of the composites, which could explain some of the trends obtained in the DMA tests of the composites.
Figure 9 shows the bottom view of the composite shown in
Figure 8F at the same magnification. A comparison of
Figure 8F and
Figure 9 reveals the aforementioned uneven vertical fiber distribution. Indeed, a higher number of fibers can be visualized throughout the matrix in
Figure 9 in comparison to
Figure 8F. In
Figure 9, the horizontal distribution is comparable to that observed in
Figure 8F, with an overall satisfactory distribution of unbundled, interspaced fibers.
Figure 9.
Scanning electron micrograph of the bottom view of PUBIMO-11.3-20 at 45× (the scale bar corresponds to 100 µm).
Figure 9.
Scanning electron micrograph of the bottom view of PUBIMO-11.3-20 at 45× (the scale bar corresponds to 100 µm).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




