
Natural Deposit Coatings on Steel during Cathodic Protection and Hydrogen Ingress



Abstract
:1. Introduction
2. Experimental Section

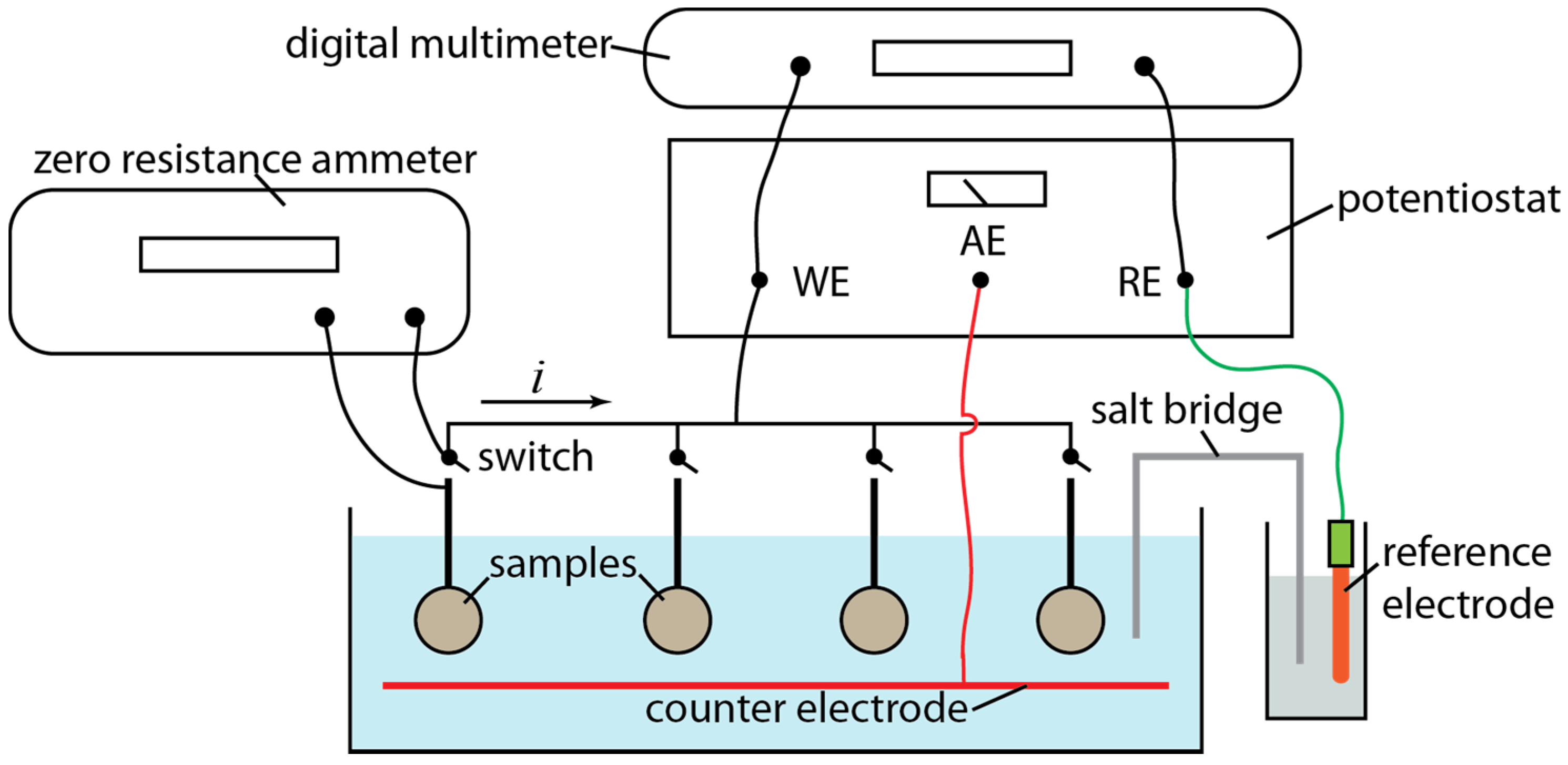{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanical Properties | Charpy V-notch Parameters | |||||
|---|---|---|---|---|---|---|
| Yield Stress (N/mm2) | Tensile strength (N/mm2) | Elongation (%) | Reduction of Area (%) | Temp. (°C) | Av. Energy (J) | Single Energy (J) |
| 760 | 1000 | 12 | 50 | −20 | 58 | 44 |



3. Results and Discussion
3.1. Visual Observations

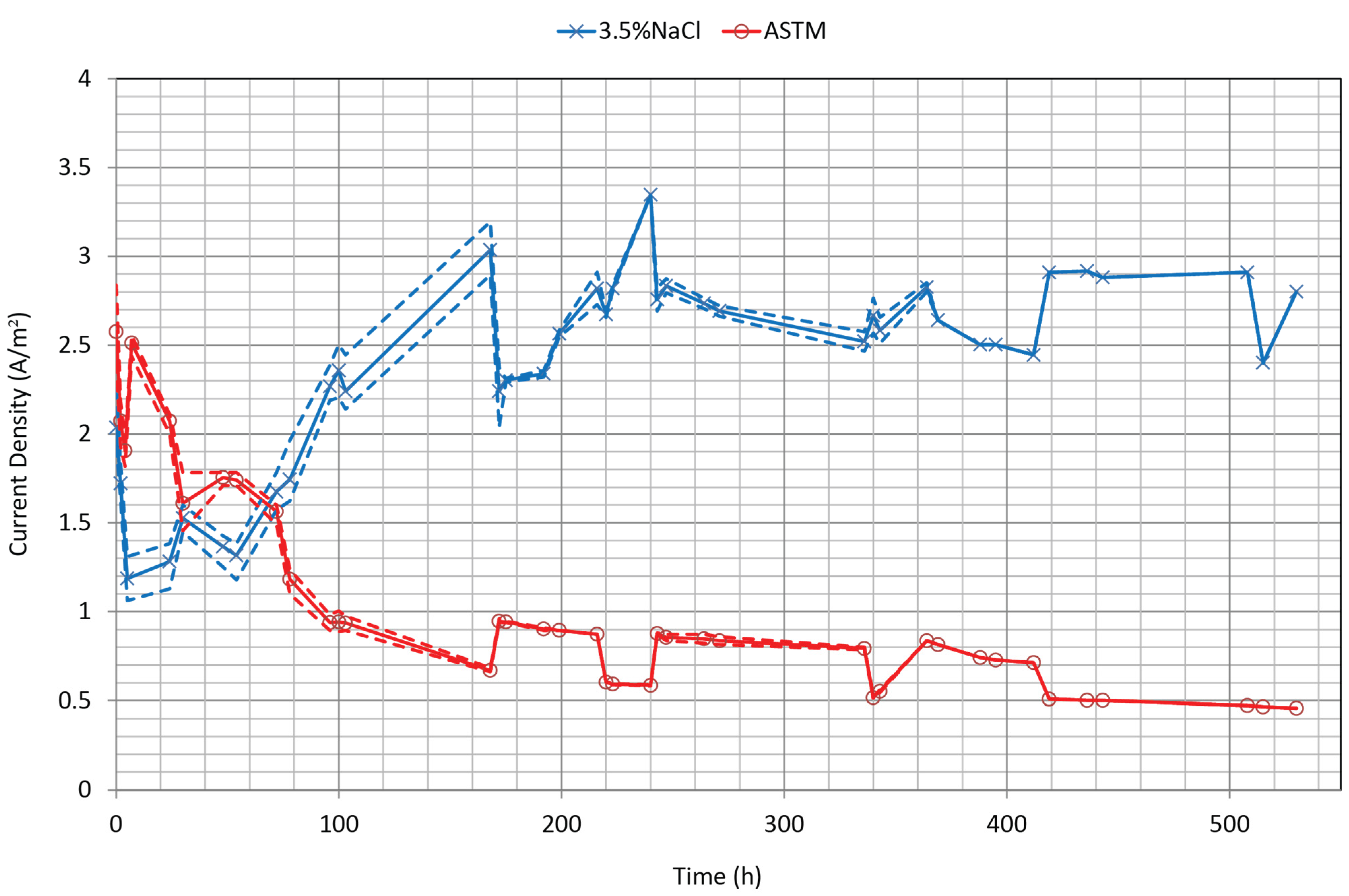
3.2. Current Density
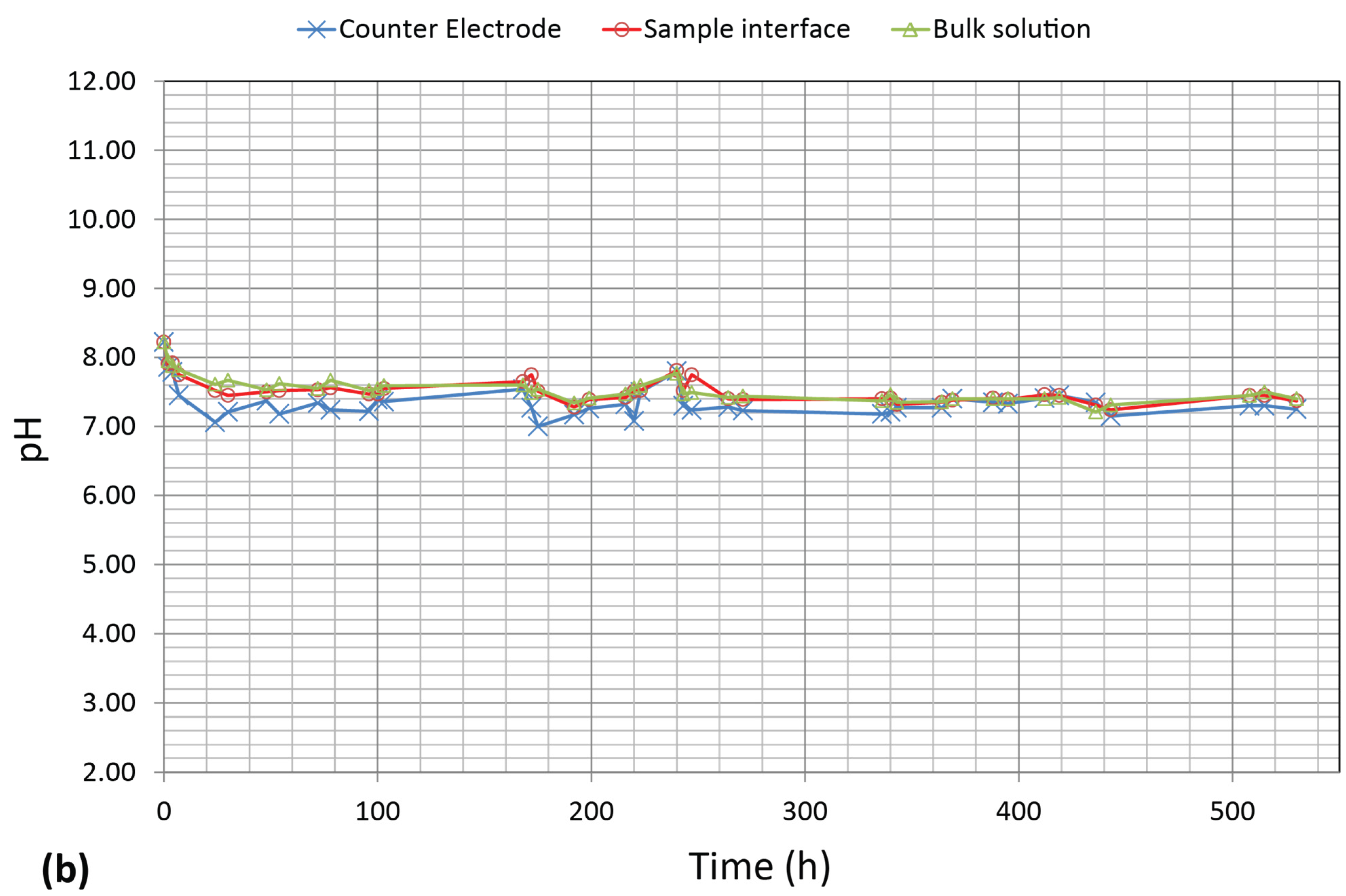
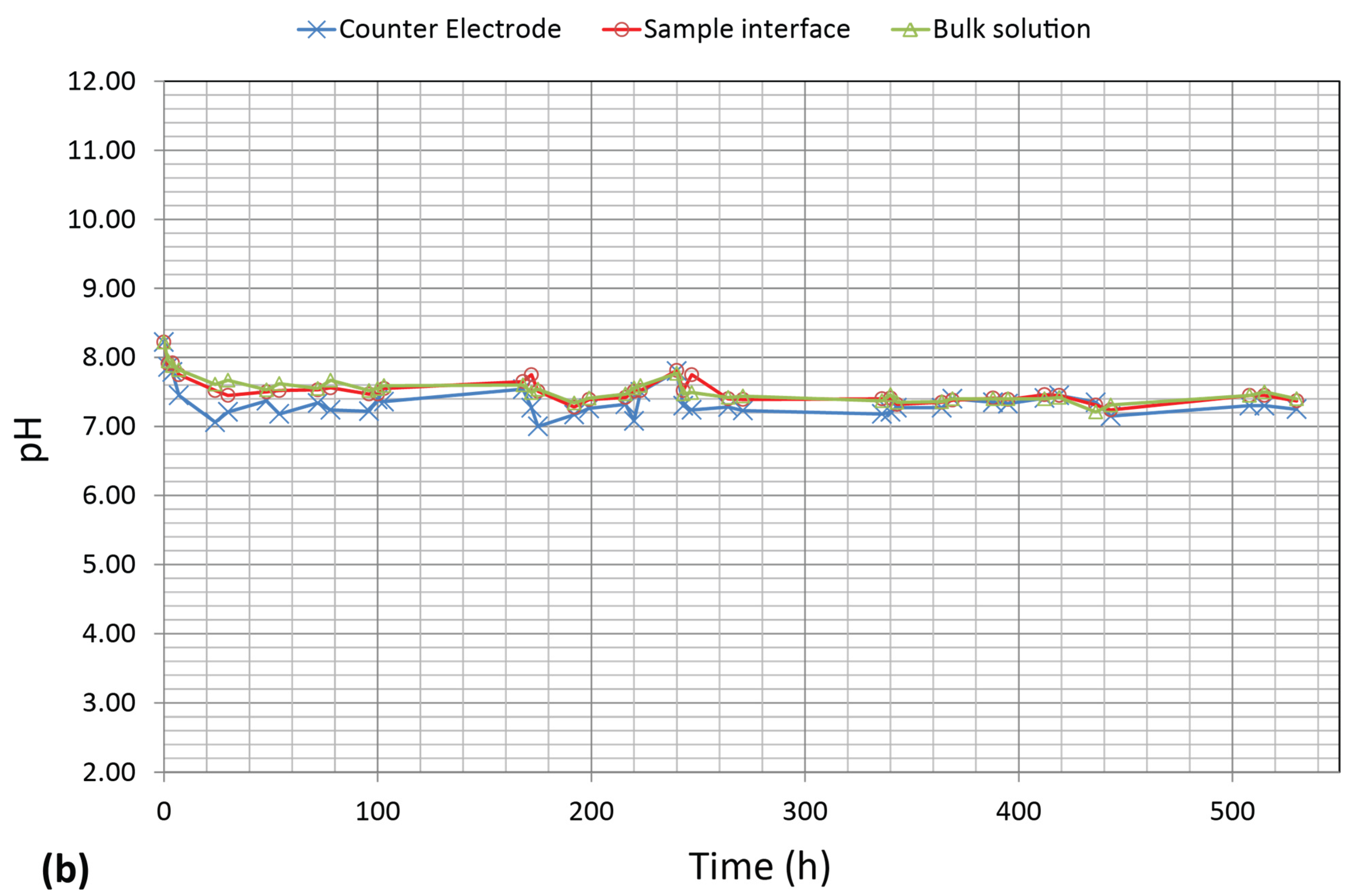
3.3. pH Measurements


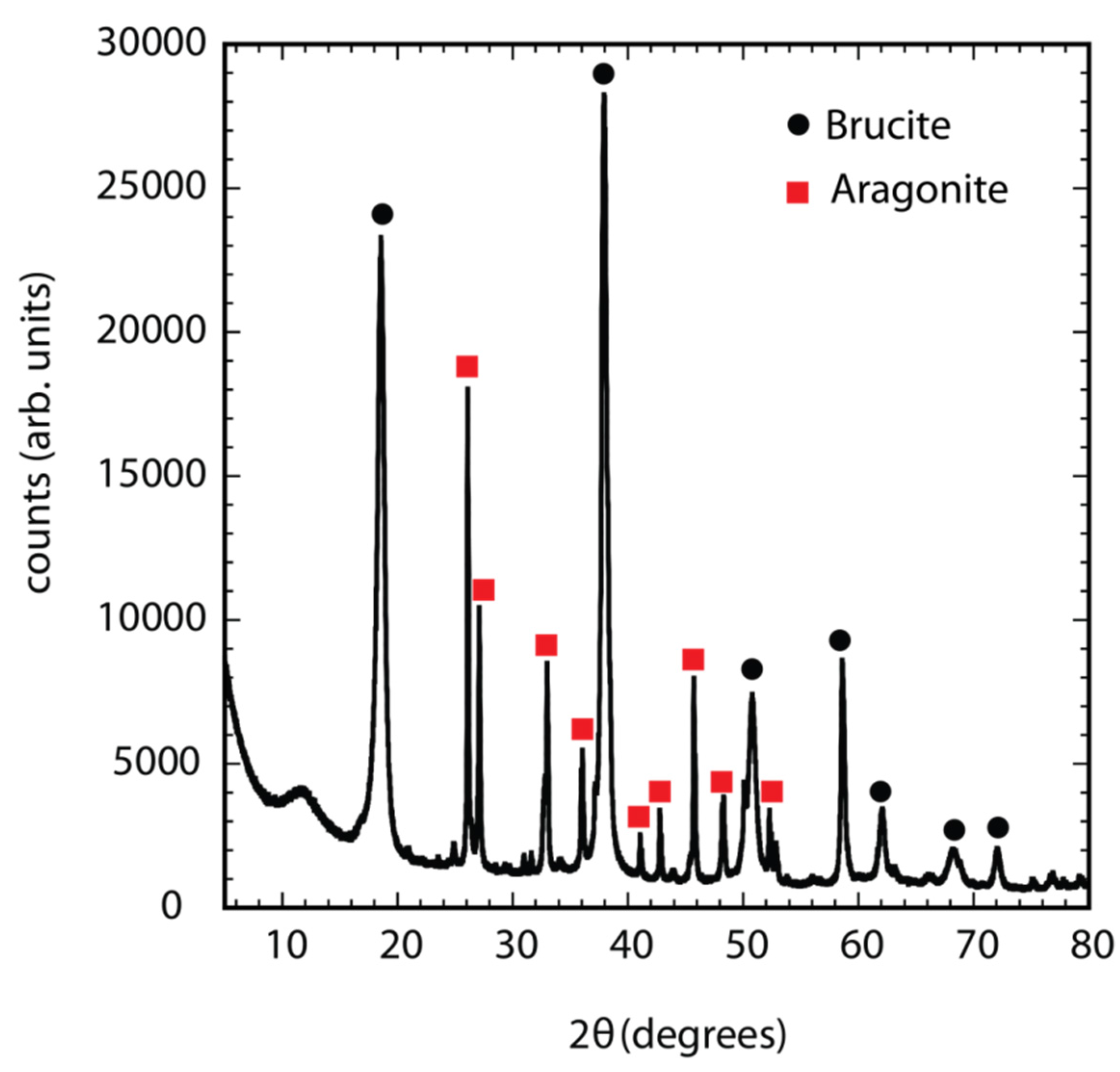
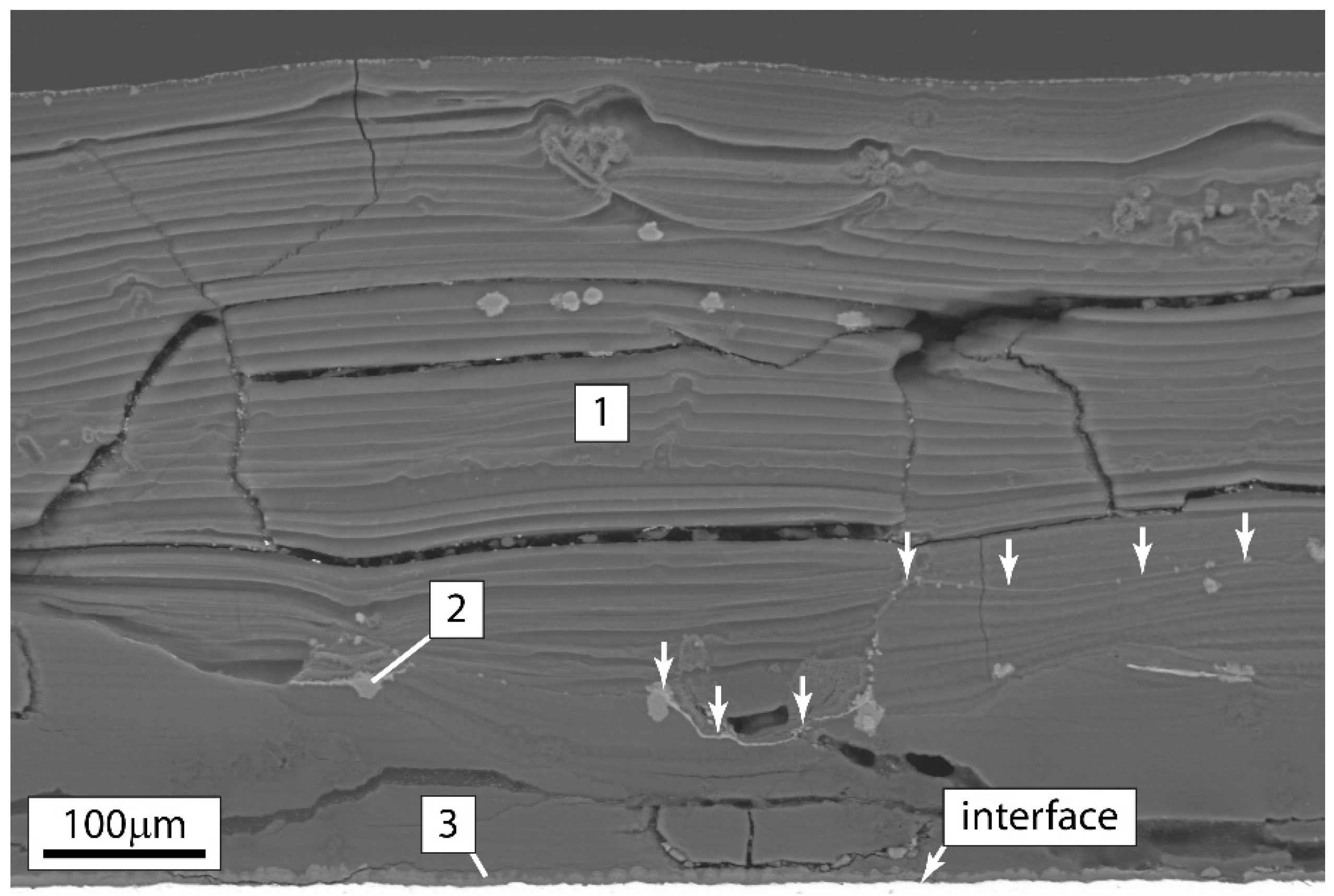
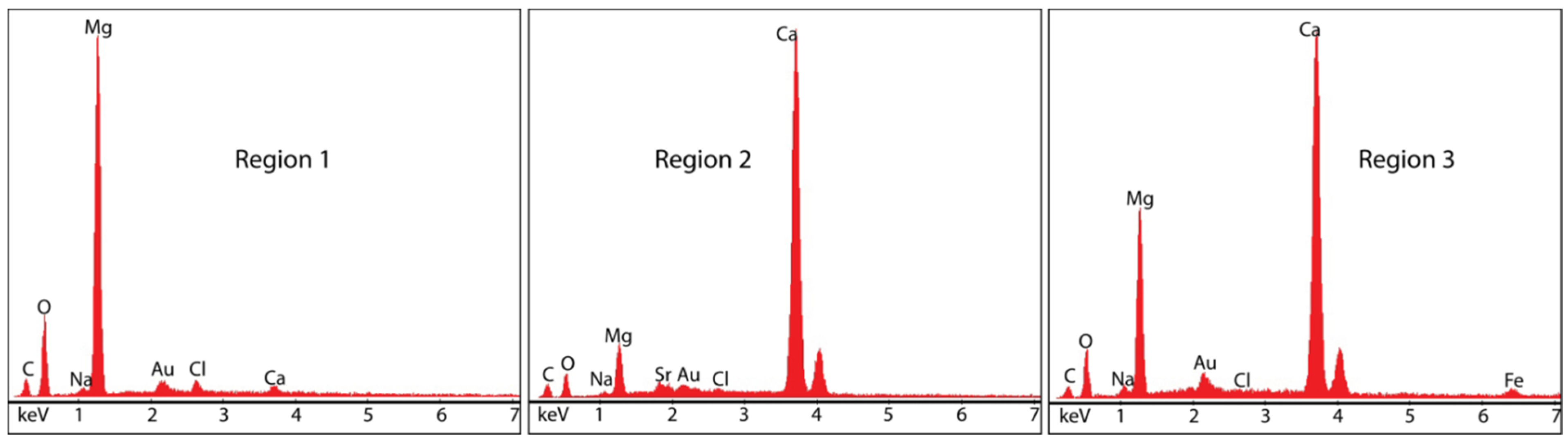
3.4. Composition

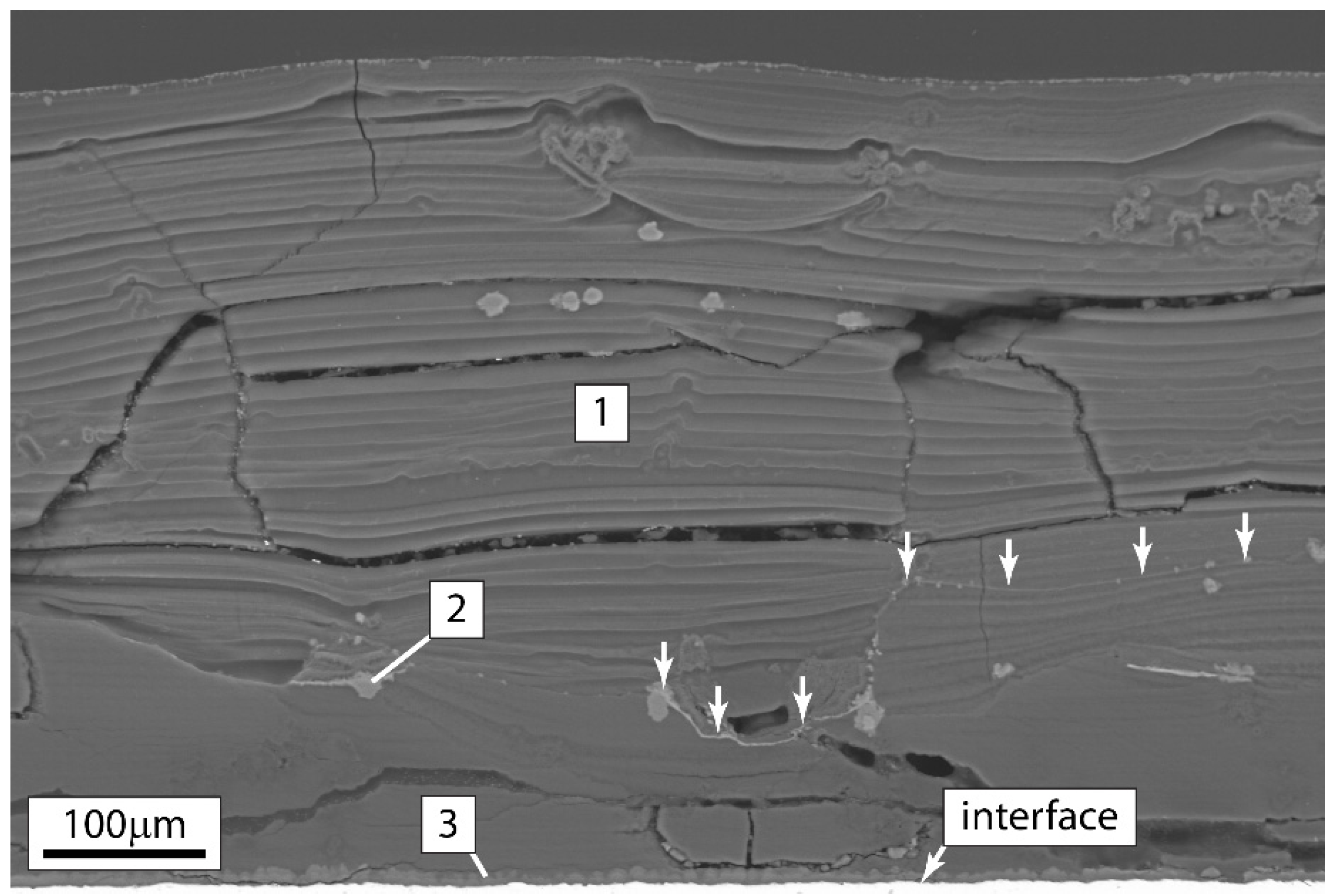

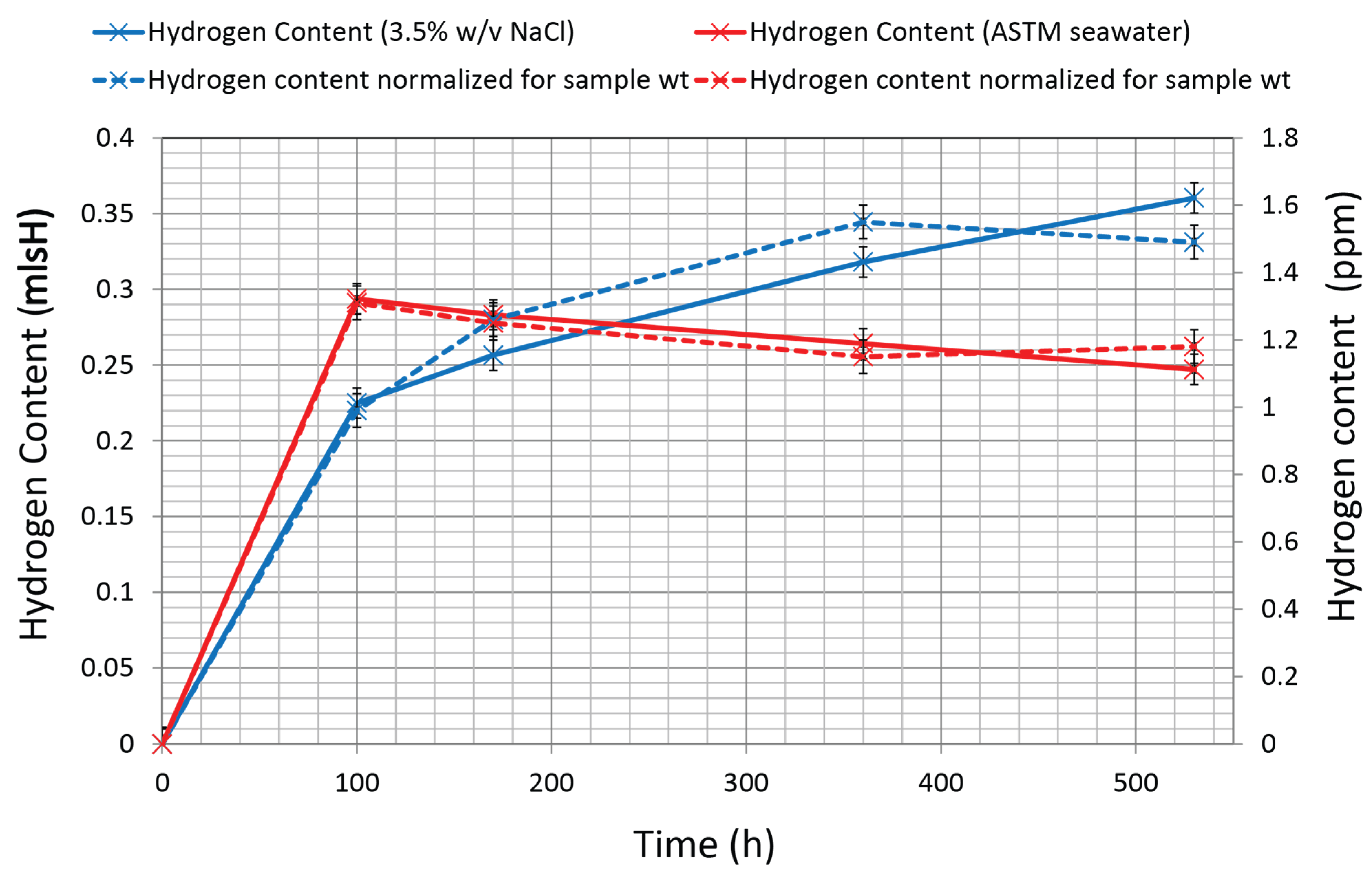
3.5. Hydrogen Content
| Time Exposure (h) | Sample ID | Weight (g) | Diffusible Hydrogen Content (mlsH) | Diffusible Hydrogen Content Normalized by wt (ppm) |
|---|---|---|---|---|
| 3.5% w/v NaCl solution | ||||
| 100 | RN4 | 20.2807 | 0.2249 | 0.99 |
| 170 | RN3 | 18.1776 | 0.2565 | 1.26 |
| 360 | RN2 | 18.3246 | 0.3181 | 1.55 |
| 530 | RN1 | 21.5952 | 0.3604 | 1.49 |
| ASTM synthetic seawater | ||||
| 100 | RS4 | 20.0166 | 0.2937 | 1.31 |
| 170 | RS3 | 20.2286 | 0.2832 | 1.25 |
| 360 | RS2 | 20.5158 | 0.2642 | 1.15 |
| 530 | RS1 | 18.6983 | 0.2471 | 1.18 |

4. Conclusions
- A more conservative approach would be to pre-charge with hydrogen in 3.5% w/v NaCl solutions over the longer term.
- The ingress of hydrogen takes place within the initial 100 h of CP when a coating is precipitated whilst it continues in uncoated samples for >360 h. The overall ingress of hydrogen is reduced when a predominantly brucite deposit is formed
- A predominantly brucite layer with some aragonite and calcite deposit forms at −1.1 V (SCE) at 20 °C in quiescent conditions.
- The coating formed consists of multiple layers of brucite interspersed with minor calcium carbonate deposits, and the initial fine layer formed is of mixed composition with slightly higher levels of Ca and Mg.
- The current density requirements for the predominantly brucite deposit are comparable with those for a mixed deposit.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wyatt, B.S. Cathodic protection of offshore structures. Anti-Corros. Methods Mater. 1977, 24, 5–10. [Google Scholar] [CrossRef]
- Elbiek, E.; Tseung, A.C.C.; Mackay, A.L. The formation of calcareous deposits during the corrosion of mild steel in seawater. Corros. Sci. 1984, 26, 669–680. [Google Scholar] [CrossRef]
- Hilbertz, W.H. Electrodeposition of minerals in sea water: Experiments and applications. IEEE J. Ocean. Eng. 1979, 4, 94–113. [Google Scholar] [CrossRef]
- Barchiche, C.; Deslouis, C.; Festy, D.; Gil, O.; Refait, P.; Touzain, S.; Tribollet, B. Characterization of calcareous deposits in artificial seawater by impedance techniques—II. Deposit of CaCO3 in the presence of Mg(II). Electrochim. Acta 2003, 48, 1645–1654. [Google Scholar] [CrossRef]
- Cottis, R.A.; Tu, K. Controlling the Components of calcareous Deposit to Protect Steel Structures from ALWC with DC and Pulse Current. In Proceedings of the NACE Corrosion Conference, Atlanta, GA, USA, 22–26 March 2009.
- Yang, Y.; Scantlebury, J.D.; Koroleva, E.V. A study of calcareous deposits on cathodically protected mild steel in artificial seawater. Metals 2015, 5, 439–456. [Google Scholar] [CrossRef]
- Hartt, W.H.; Smith, S.W. Chemistry-Structure-Property Interrelayionships for Calcareous Deposits as Stand-alone Coatings; Center for Marine Materials, Office of Naval Research: Arlington, VA, USA, 1989. [Google Scholar]
- Bockris, J.O’M. Hydrogen. Materials 2011, 4, 2073–2091. [Google Scholar] [CrossRef]
- Wolfson, S.L.; Hartt, W.H. An initial investigation of calcareous deposits upon cathodic steel surfaces in sea water. Corrosion 1981, 37, 70–76. [Google Scholar] [CrossRef]
- Deslouis, C.; Festy, D.; Gil, O.; Maillot, V.; Touzain, S.; Tribollet, B. Characterization of calcareous deposits in artificial sea water by impedances techniques: 2-deposit of Mg(OH)2 without CaCO3. Electrochim. Acta 2000, 45, 1837–1845. [Google Scholar] [CrossRef]
- Akamine, K.; Kashiki, I. Corrosion protection of steel by calcareous electrodeposition in seawater: Part 1: Mechanism of electrodeposition. Eng. Rev. 2002, 36, 636–642. [Google Scholar]
- D1141–98 Standard Practice for the Preparation of Substitute Ocean Water; American Society for Testing and Materials (ASTM): West Conshohocken, PA, USA, 2013.
- Engell, H.J.; Forchhammer, P. The change of pH under a paint film due to cathodic protection. Corros. Sci. 1965, 5, 479–484. [Google Scholar] [CrossRef]
- Luo, J.S.; Lee, R.U.; Chen, T.Y.; Hartt, W.H.; Smith, S.W. Formation of calcareous deposits under different modes of cathodic polarization. Corrosion 1991, 47, 189–196. [Google Scholar] [CrossRef]
- Okstad, T.; Rannestad, O.; Johnsen, R.; Nisancioglu, K. Significance of hydrogen evolution during cathodic protection of carbon steel in seawater. Corrosion 2007, 63, 857–865. [Google Scholar] [CrossRef]
- Neville, A.; Morizot, A.P. Calcareous scales formed by cathodic protection—An assessment of characteristics and kinetics. J. Cryst. Growth 2002, 243, 490–502. [Google Scholar] [CrossRef]
- Kunjapur, M.M.; Hartt, W.H.; Smith, S.W. Influence of Temperature and Exposure Time upon Calcareous Deposits. Corrosion 1987, 43, 674–679. [Google Scholar] [CrossRef]
- Lin, S.; Dexter, S.C. Effects of temperature and magnesium ions on calcareous deposition. Corros. J. 1988, 44, 615–622. [Google Scholar] [CrossRef]
- Barchiche, C.; Deslouis, C.; Gil, O.; Refait, P.; Tribollet, B. Characterization of calcareous deposits by electrochemical methods: Role of sulphates, calcium concentration and temperature. Electrochim. Acta 2004, 49, 2833–2839. [Google Scholar] [CrossRef]
- Barchiche, C.; Deslouis, C.; Gil, O.; Joiret, S.; Refait, P.; Tribollet, B. Role of sulphate ions on the formation of calcareous deposits on steel in artificial seawater; the formation of Green Rust compounds during cathodic protection. Electrochim. Acta 2009, 54, 3580–3588. [Google Scholar] [CrossRef]
- Deslouis, C.; Festy, D.; Gil, O. Characterization of calcareous deposits in artificial seawater by impedance techniques—I. Deposit of CaCO3 in the absence of Mg(OH)2. Electrochim. Acta 1998, 43, 1891–1901. [Google Scholar] [CrossRef]
- Leeds, S.S.; Cottis, R.A. An Investigation into the Influence of Surface Films on the Mechanism of Cathodic Protection. In Proceedings of the NACE Corrosion Conference, San Diego, CA, USA, 12–16 March 2006.
- Fairhurst, D. Offshore cathodic protection. What we have learnt? J. Corros. Sci. Eng. 2003, 4, 1–17. [Google Scholar]
- Ju, C.P.; Don, J.; Rigsbee, J.M. A high voltage electron microscopy study of hydrogen-induced damage in a low alloy. Medium Carbon Steel Mater. Sci. Eng. 1986, 77, 115–123. [Google Scholar]
- Thomas, R.L.S.; Scully, J.R.; Gangloff, R.P. Internal hydrogen embrittlement of ultrahigh-strength AERMET 100 steel. Metall. Mater. Trans. A 2003, 34, 327–344. [Google Scholar]
- Gangloff, R.P. Hydrogen Assisted Cracking of High Strength Alloys. In Comprehensive Structural Integrity, Elsevier Science; Milne, I., Ritchie, R.O., Karihaloo, B.L., Eds.; Elsevier: Amsterdam, The Netherlands; Boston, MA, USA, 2003; Volume 6, p. 31. [Google Scholar]
- Murakami, Y.; Kanezaki, T.; Mine, Y. Hydrogen effect against hydrogen embrittlement. Metall. Mater. Trans. A 2010, 41, 2548–2562. [Google Scholar] [CrossRef]
- Hinds, G.; Turnbull, A. Technical note: Does calcareous scale formation on cathodically protected steel affect hydrogen uptake? Corrosion 2005, 61, 835–837. [Google Scholar] [CrossRef]
- Hartt, W.H. Discussion: Does calcareous scale formation on cathodically protected steel affect hydrogen uptake? Corrosion 2006, 62, 947–949. [Google Scholar] [CrossRef]
- Ou, K.C.; Wu, J.K. Effect of calcareous deposits formation on the hydrogen absorption of steel. Mater. Chem. Phys. 1997, 48, 52–55. [Google Scholar] [CrossRef]
- Olsen, S.; Hesjevik, S.M. Hydrogen Embrittlement from CP on Supermartensitic Stainless Steels—Recommendations for New Qualification Methods. In Proceedings of the NACE Corrosion Conference, New Orleans, LA, USA, March 2004.
- Offshore Mooring Chain; Offshore Standard DNV-OS-E302; Det Norske Veritas (DNV GL): Oslo, Norway, 2013.
- Hack, H.P.; Guanti, R.J. Effect of High Flow on Calcareous Deposits and Cathodic Protection Current Density; Research and Development Report, DTRC/SME-87/82; David Taylor Research Center: Bethesda, MD, USA, 1988. [Google Scholar]
© 2015 by W.R. Smith and TWI Ltd.; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, W.R.; Paul, S. Natural Deposit Coatings on Steel during Cathodic Protection and Hydrogen Ingress. Coatings 2015, 5, 816-829. https://doi.org/10.3390/coatings5040816
Smith WR, Paul S. Natural Deposit Coatings on Steel during Cathodic Protection and Hydrogen Ingress. Coatings. 2015; 5(4):816-829. https://doi.org/10.3390/coatings5040816
Chicago/Turabian StyleSmith, Wayne R., and Shiladitya Paul. 2015. "Natural Deposit Coatings on Steel during Cathodic Protection and Hydrogen Ingress" Coatings 5, no. 4: 816-829. https://doi.org/10.3390/coatings5040816
APA StyleSmith, W. R., & Paul, S. (2015). Natural Deposit Coatings on Steel during Cathodic Protection and Hydrogen Ingress. Coatings, 5(4), 816-829. https://doi.org/10.3390/coatings5040816