Investigations on Forming Ether Coated Iron Nanoparticle Materials by First-Principle Calculations and Molecular Dynamic Simulations

Abstract
:1. Introduction
2. Methods
2.1. MD Simulation and ReaxFF Force Field
2.2. MD Simulation Systems and Setup
2.3. First-Principle Calculations
3. Results and Discussion
3.1. First-Principle Calculations
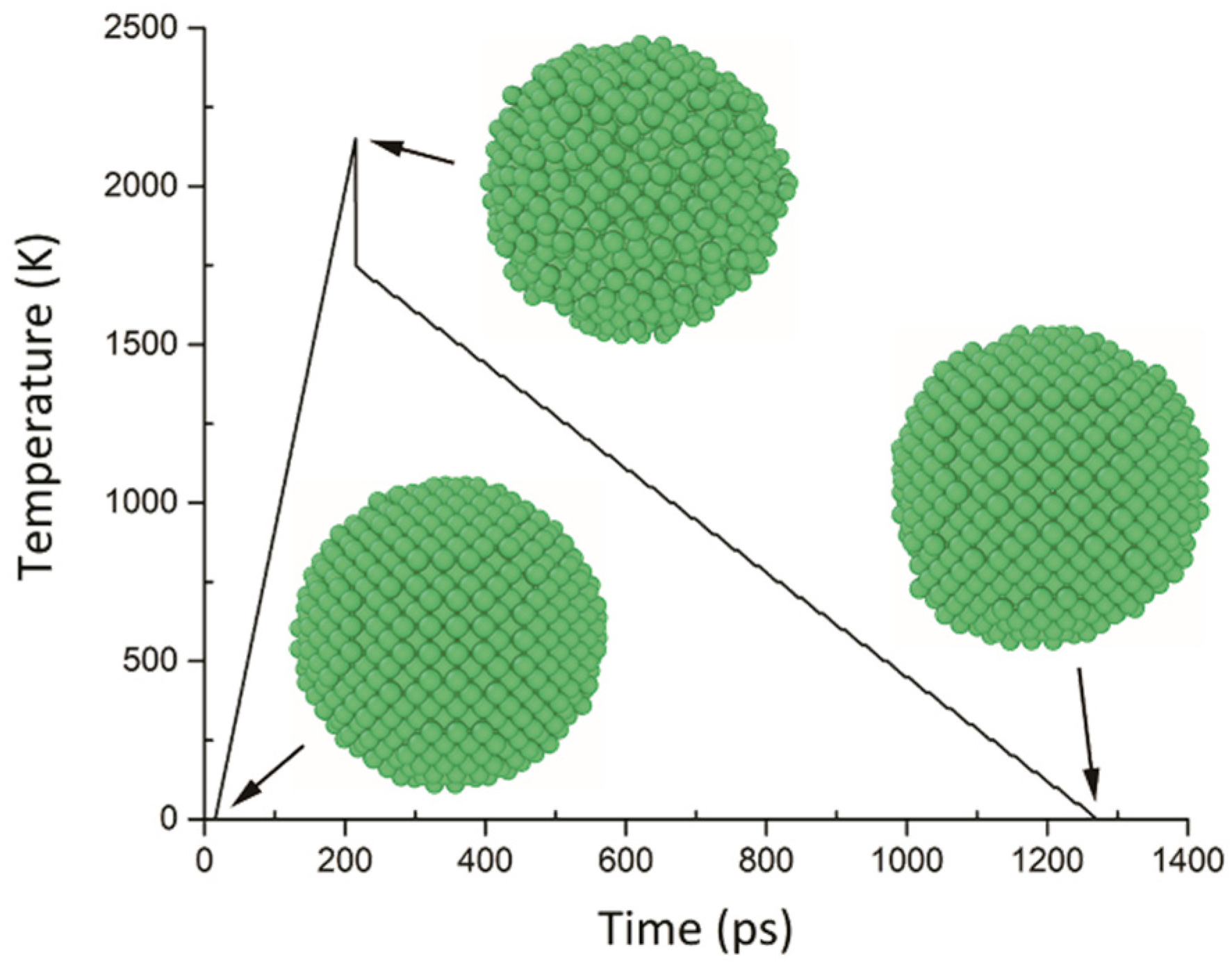
3.2. Single Sphere Coating Model
3.3. Double Sphere Coating Model
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kabir, E.; Kumar, V.; Kim, K.H.; Yip, A.C.; Sohn, J.R. Environmental impacts of nanomaterials. J. Environ. Manag. 2018, 225, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.P.; Li, X.Q.; Cao, J.; Zhang, W.X.; Wang, H.P. Characterization of zero-valent iron nanoparticles. Adv. Colloid Interface Sci. 2006, 120, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.B.; Zhang, W.X. Synthesizing nanoscale iron particles for rapid and complete dechlorination of TCE and PCBs. Environ. Sci. Technol. 1997, 31, 2154–5156. [Google Scholar] [CrossRef]
- Huber, D.L. Synthesis, properties, and applications of iron nanoparticles. Small 2005, 1, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Newsome, L.; Morris, K.; Cleary, A.; Masters-Waage, N.K.; Boothman, C.; Joshi, N.; Atherton, A.; Lloyd, J.R. The impact of iron nanoparticles on technetium-contaminated groundwater and sediment microbial communities. J. Hazard. Mater. 2019, 364, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, M.; Ebrahimi-Nik, M.; Heidari, A.; Abbaspour-Fard, M.H. Improvement of biogas production from slaughterhouse wastewater using biosynthesized iron nanoparticles from water treatment sludge. Renew. Energy 2019, 135, 496–501. [Google Scholar] [CrossRef]
- Eremin, A.; Gurentsov, E. Evaporation temperature depression with decrease of iron nanoparticle size. Validation of semi-empirical models. Mater. Chem. Phys. 2019, 228, 180–186. [Google Scholar] [CrossRef]
- Yadav, A.; Kumar, R.; Choudhary, H.K.; Sahoo, B. Graphene-oxide coating for corrosion protection of iron particles in saline water. Carbon 2018, 140, 477–487. [Google Scholar] [CrossRef]
- Leng, Y.; Sato, K.; Li, J.G.; Ishigaki, T.; Iijima, M.; Kamiya, H.; Yoshida, T. Iron nanoparticles dispersible in both ethanol and water for direct silica coating. Powder Technol. 2009, 196, 80–84. [Google Scholar] [CrossRef]
- Dulinska-Molak, I.; Chlanda, A.; Li, J.; Wang, X.; Bystrzejewski, M.; Kawazoe, N.; Chen, C.; Swieszkowski, W. The influence of carbon-encapsulated iron nanoparticles on elastic modulus of living human mesenchymal stem cells examined by atomic force microscopy. Micron 2018, 108, 41–48. [Google Scholar] [CrossRef]
- Khramtsov, P.; Kropaneva, M.; Byzov, I.Y.; Minin, A.; Mysik, A.; Timganova, V.; Bochkova, M.; Uimin, M.; Zamorina, S.; Yermakov, A.; et al. Conjugation of carbon coated-iron nanoparticles with biomolecules for NMR-based assay. Colloids Surf. B Biointerfaces 2019, 176, 256–264. [Google Scholar] [CrossRef] [PubMed]
- McGrath, A.J.; Dolan, C.; Cheong, S.; Herman, D.A.; Naysmith, B.; Zong, F.; Galvosas, P.; Farrand, K.J.; Hermans, I.F.; Brimble, M.; et al. Stability of polyelectrolyte-coated iron nanoparticles for T2-weighted magnetic resonance imaging. J. Magn. Magn. Mater. 2017, 439, 251–258. [Google Scholar] [CrossRef]
- Hong, S.; van Duin, A.C. Atomistic-scale analysis of carbon coating and its effect on the oxidation of aluminum nanoparticles by ReaxFF-molecular dynamics simulations. J. Phys. Chem. C 2016, 120, 9464–9474. [Google Scholar] [CrossRef]
- Yi, Q.; Xu, J.; Liu, Y.; Zhai, D.; Zhou, K.; Pan, D. Molecular dynamics study on core-shell structure stability of aluminum encapsulated by nano-carbon materials. Chem. Phys. Lett. 2017, 669, 192–195. [Google Scholar] [CrossRef]
- Kim, K.T.; Kim, D.W.; Kim, S.H.; Kim, C.K.; Choi, Y.J. Synthesis and improved explosion behaviors of aluminum powders coated with nano-sized nickel film. Appl. Surf. Sci. 2017, 415, 104–108. [Google Scholar] [CrossRef]
- Sun, R.; Liu, P.; Qi, H.; Liu, J.; Ding, T. Molecular dynamic simulations of ether-coated aluminum nano-particles as a novel hydrogen source. J. Nanopart. Res. 2019, 21, 72. [Google Scholar] [CrossRef]
- Liu, J.; Liu, P.; Wang, M. Molecular dynamics simulations of aluminum nanoparticles adsorbed by ethanol molecules using the ReaxFF reactive force field. Comput. Mater. Sci. 2018, 151, 95–105. [Google Scholar] [CrossRef]
- Zhang, Y.R.; van Duin, A.C.; Luo, K.H. Investigation of ethanol oxidation over aluminum nanoparticle using ReaxFF molecular dynamics simulation. Fuel 2018, 234, 94–100. [Google Scholar] [CrossRef]
- Pašti, I.A.; Gavrilov, N.M.; Mentus, S.V. Hydrogen adsorption on palladium and platinum overlayers: DFT study. Adv. Phys. Chem. 2011, 2011, 1–8. [Google Scholar] [CrossRef]
- Xie, W.; Peng, L.; Peng, D.; Gu, F.L.; Liu, J. Processes of H2 adsorption on Fe (110) surface: A density functional theory study. Appl. Surf. Sci. 2014, 296, 47–52. [Google Scholar] [CrossRef]
- Chiang, H.N.; Nachimuthu, S.; Cheng, Y.C.; Damayanti, N.P.; Jiang, J.C. A DFT study of ethanol adsorption and decomposition on α-Al2O3 (0001) surface. Appl. Surf. Sci. 2016, 363, 636–643. [Google Scholar] [CrossRef]
- Tan, X.; Zhou, J.; Peng, Y. First-principles study of oxygen adsorption on Fe (110) surface. Appl. Surf. Sci. 2012, 258, 8484–8491. [Google Scholar] [CrossRef]
- Ossowski, T.; Kiejna, A. Oxygen adsorption on Fe (110) surface revisited. Surf. Sci. 2015, 637, 35–41. [Google Scholar] [CrossRef]
- Fatti, G.; Restuccia, P.; Calandra, C.; Righi, M.C. Phosphorus adsorption on Fe (110): An ab initio comparative study of iron passivation by different adsorbates. J. Phys. Chem. C 2018, 122, 28105–28112. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Tamijani, A.A.; Salam, A.; de Lara-Castells, M.P. Adsorption of noble-gas atoms on the TiO2 (110) surface: An ab initio-assisted study with van der Waals-corrected DFT. J. Phys. Chem. C 2016, 120, 18126–18139. [Google Scholar] [CrossRef]
- Thonhauser, T.; Cooper, V.R.; Li, S.; Puzder, A.; Hyldgaard, P.; Langreth, D.C. Van der Waals density functional: Self-consistent potential and the nature of the van der Waals bond. Phys. Rev. B 2007, 76, 125112. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Hemeda, A.A.; Esteves, R.J.A.; McLeskey Jr, J.T.; Gad-el-Hak, M.; Khraisheh, M.; Tafreshi, H.V. Molecular dynamic simulations of fibrous distillation membranes. Int. Commun. Heat Mass Transf. 2018, 98, 304–309. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Bou-Rabee, N. Time integrators for molecular dynamics. Entropy 2013, 16, 138–162. [Google Scholar] [CrossRef]
- in’t Veld, P.J.; Plimpton, S.J.; Grest, G.S. Accurate and efficient methods for modeling colloidal mixtures in an explicit solvent using molecular dynamics. Comput. Phys. Commun. 2008, 179, 320–329. [Google Scholar] [CrossRef]
- Tersoff, J. New empirical model for the structural properties of silicon. Phys. Rev. Lett. 1986, 56, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guo, X. ReaxFF molecular dynamics simulation of pyrolysis and combustion of pyridine. Fuel Process. Technol. 2017, 161, 107–115. [Google Scholar] [CrossRef]
- Mortier, W.J.; Ghosh, S.K.; Shankar, S. Electronegativity-equalization method for the calculation of atomic charges in molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320. [Google Scholar] [CrossRef]
- Nayebi, P.; Zaminpayma, E. A molecular dynamic simulation study of mechanical properties of graphene–polythiophene composite with Reax force field. Phys. Lett. A 2016, 380, 628–633. [Google Scholar] [CrossRef]
- Aryanpour, M.; Van Duin, A.C.; Kubicki, J.D. Development of a reactive force field for iron-oxyhydroxide systems. J. Phys. Chem. A 2010, 114, 6298–6307. [Google Scholar] [CrossRef]
- Russo Jr, M.F.; Li, R.; Mench, M.; Van Duin, A.C. Molecular dynamic simulation of aluminum-water reactions using the ReaxFF reactive force field. Int. J. Hydrogen Energy 2011, 36, 5828–5835. [Google Scholar] [CrossRef]
- Hong, S.; Van Duin, A.C. Molecular dynamics simulations of the oxidation of aluminum nanoparticles using the ReaxFF reactive force field. J. Phys. Chem. C 2015, 119, 17876–17886. [Google Scholar] [CrossRef]
- Liu, P.; Liu, J.; Wang, M. Ignition and combustion of nano-sized aluminum particles: A reactive molecular dynamics study. Combust. Flame 2019, 201, 276–289. [Google Scholar] [CrossRef]
- Duy, T.P.; Van Hoang, V. Atomic mechanism of homogeneous melting of bcc Fe at the limit of superheating. Phys. B 2012, 407, 978–984. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 2384, 2384–2393. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The open visualization tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Milman, V.; Refson, K.; Clark, S.J.; Pickard, C.J.; Yates, J.R.; Gao, S.P.; Hasnip, P.J.; Probert, M.I.J.; Perlov, A.; Segall, M.D. Electron and vibrational spectroscopies using DFT, plane waves and pseudopotentials: CASTEP implementation. J. Mol. Struct. THEOCHEM 2010, 954, 22–35. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and opensource software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Henz, B.J.; Hawa, T.; Zachariah, M. Molecular dynamics simulation of the energetic reaction between Ni and Al nanoparticles. J. Appl. Phys. 2009, 105, 124310. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms | Charge (Multiple of Charge) |
|---|---|
| H1 | 0.2024 |
| H2 | 0.2029 |
| H3 | 0.2029 |
| H4 | 0.162 |
| H5 | 0.162 |
| C1 | −0.454 |
| C2 | −0.0303 |
| O1 | 0.3535 |
| Adsorption Sites | Adsorption Energy (kcal/mol) |
|---|---|
| atop | −20.339 |
| hcp | −19.500 |
| long bridge | −24.307 |
| short bridge | −20.453 |
| Adsorption Sites | C–C | C–O | C–H | ˂C–O–C | ˂O–C–H | ˂H10–O4–H9 |
|---|---|---|---|---|---|---|
| Original | 1.513 | 1.427 | 1.109 | 112.742 | 109.755 | 108.272 |
| atop | 1.497 | 1.44 | 1.107 | 108.843 | 108.938 | 107.123 |
| hcp | 1.498 | 1.437 | 1.106 | 109.887 | 109.056 | 105.424 |
| long bridge | 1.5 | 1.434 | 1.106 | 110.962 | 109.656 | 106.749 |
| short bridge | 1.5 | 1.435 | 1.103 | 110.424 | 109.087 | 105.562 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Hui, S.; Liu, P.; Sun, R.; Wang, M. Investigations on Forming Ether Coated Iron Nanoparticle Materials by First-Principle Calculations and Molecular Dynamic Simulations. Coatings 2019, 9, 395. https://doi.org/10.3390/coatings9060395
Sun J, Hui S, Liu P, Sun R, Wang M. Investigations on Forming Ether Coated Iron Nanoparticle Materials by First-Principle Calculations and Molecular Dynamic Simulations. Coatings. 2019; 9(6):395. https://doi.org/10.3390/coatings9060395
Chicago/Turabian StyleSun, Junlei, Shixuan Hui, Pingan Liu, Ruochen Sun, and Mengjun Wang. 2019. "Investigations on Forming Ether Coated Iron Nanoparticle Materials by First-Principle Calculations and Molecular Dynamic Simulations" Coatings 9, no. 6: 395. https://doi.org/10.3390/coatings9060395