Making a Short Story Long: Regulation of P-TEFb and HIV-1 Transcriptional Elongation in CD4+ T Lymphocytes and Macrophages

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Transcriptional Elongation
3. A Perspective on P-TEFb
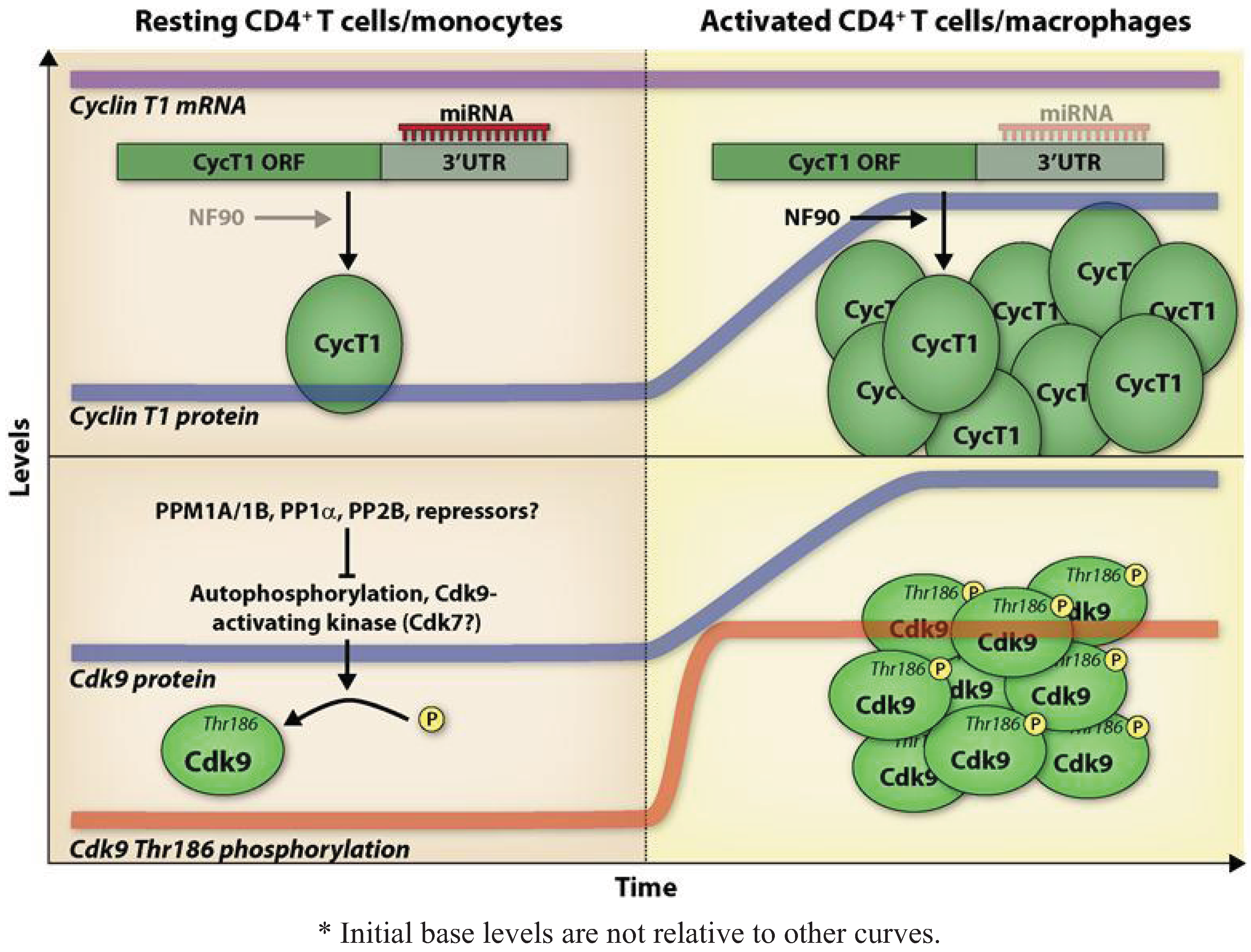
4. Expression and Regulation of P-TEFb in CD4+ T Cells and Macrophages Affects HIV-1 Replication
4.1. Expression and Regulation of Cdk9

4.2. Expression and Regulation of Cyclin T1 and Cyclin T2
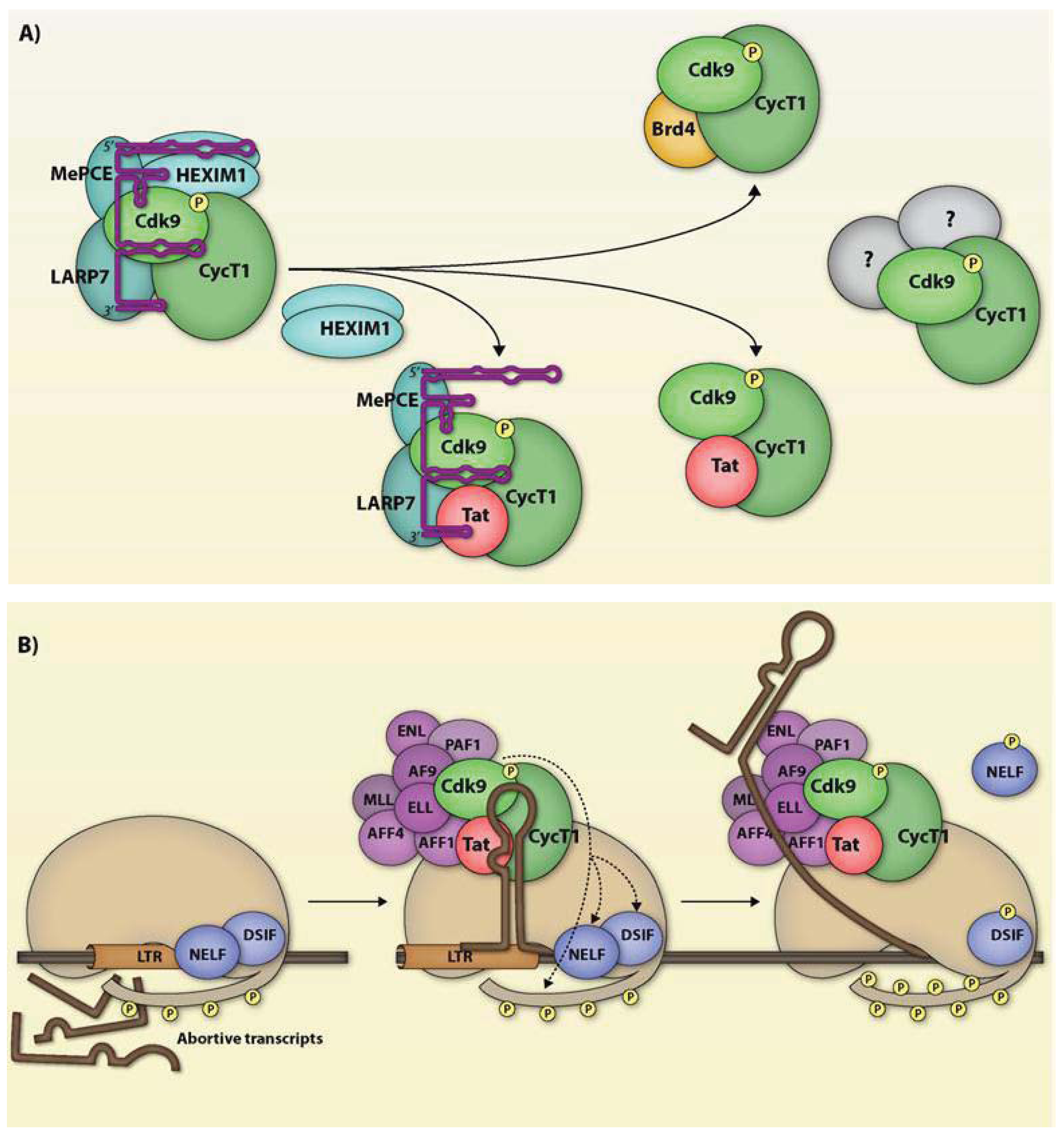
5. The ‘Complex’ Diversity of P-TEFb

5.1. P-TEFb and 7SK snRNP
5.2. P-TEFb and Brd4
5.3. P-TEFb and the SEC
5.4. P-TEFb and Other Multi-Protein Complexes
6. P-TEFb and HIV-1 Latency
Avenues for Future Research
- How many miRNAs individually and cooperatively regulate Cyclin T1 in CD4+ T cells and macrophages? What is their range of targets and how do these targets play into P-TEFb function?
- Does HIV-1 Tat utilize the novel Cdk9/Cyclin T1 complexes identified by HT-IP/MS?
- How can P-TEFb expression and function be manipulated (e.g., by inducing Cdk9 T-loop phosphorylation or using antagomiRs to inhibit Cyclin T1 responsive miRNAs) to reactivate latent provirus?
References
- van Driel, R.; Verschure, P.J. Nuclear Organization and Gene Expression: Visualization of Transcription and Higher Order Chromatin Structure; INSERM: Paris, France, 2001. [Google Scholar]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Core, L.J.; Lis, J.T. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science 2008, 319, 1791–1792. [Google Scholar] [CrossRef]
- Hargreaves, D.C.; Horng, T.; Medzhitov, R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 2009, 138, 129–145. [Google Scholar] [CrossRef]
- Sikorski, T.W.; Buratowski, S. The basal initiation machinery: Beyond the general transcription factors. Curr. Opin. Cell Biol. 2009, 21, 344–351. [Google Scholar] [CrossRef]
- Boeing, S.; Rigault, C.; Heidemann, M.; Eick, D.; Meisterernst, M. RNA polymerase II C-terminal heptarepeat domain Ser-7 phosphorylation is established in a mediator-dependent fashion. J. Biol. Chem. 2010, 285, 188–196. [Google Scholar]
- Hirose, Y.; Ohkuma, Y. Phosphorylation of the C-terminal domain of RNA polymerase II plays central roles in the integrated events of eucaryotic gene expression. J. Biochem. 2007, 141, 601–608. [Google Scholar] [CrossRef]
- Kim, H.; Erickson, B.; Luo, W.; Seward, D.; Graber, J.H.; Pollock, D.D.; Megee, P.C.; Bentley, D.L. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat. Struct. Mol. Biol. 2010, 17, 1279–1286. [Google Scholar]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef]
- Nechaev, S.; Fargo, D.C.; dos Santos, G.; Liu, L.; Gao, Y.; Adelman, K. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 2010, 327, 335–338. [Google Scholar]
- Nechaev, S.; Adelman, K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta 1809, 34–45. [Google Scholar]
- Glover-Cutter, K.; Larochelle, S.; Erickson, B.; Zhang, C.; Shokat, K.; Fisher, R.P.; Bentley, D.L. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol. Cell Biol. 2009, 29, 5455–5464. [Google Scholar] [CrossRef]
- Chen, H.; Contreras, X.; Yamaguchi, Y.; Handa, H.; Peterlin, B.M.; Guo, S. Repression of RNA polymerase II elongation in vivo is critically dependent on the C-terminus of Spt5. PLoS One 2009, 4, e6918. [Google Scholar]
- Fujita, T.; Piuz, I.; Schlegel, W. The transcription elongation factors NELF, DSIF and P-TEFb control constitutive transcription in a gene-specific manner. FEBS Lett. 2009, 583, 2893–2898. [Google Scholar] [CrossRef]
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol. Cell 2006, 21, 227–237. [Google Scholar] [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell Biol. 2004, 24, 787–795. [Google Scholar]
- Karn, J.; Stoltzfus, C.M. Transcriptional and posttranscriptional regulation of Hiv-1 gene expression. Cold Spring Harb. Perspect. Med. 2012, 2, a006916. [Google Scholar]
- Zhou, Q.; Tiandao, L.; Price, D.H. RNA Polymerase II elongation control. Annu. Rev. Biochem. 2012, 81, 14.11–14.25. [Google Scholar]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef]
- Bres, V.; Gomes, N.; Pickle, L.; Jones, K.A. A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes. Dev. 2005, 19, 1211–1226. [Google Scholar] [CrossRef]
- Ardehali, M.B.; Lis, J.T. Tracking rates of transcription and splicing in vivo. Nat. Struct. Mol. Biol. 2009, 16, 1123–1124. [Google Scholar] [CrossRef]
- Maiuri, P.; Knezevich, A.; de Marco, A.; Mazza, D.; Kula, A.; McNally, J.G.; Marcello, A. Fast transcription rates of RNA polymerase II in human cells. EMBO Rep. 2011, 12, 1280–1285. [Google Scholar] [CrossRef]
- Bres, V.; Yoh, S.M.; Jones, K.A. The multi-tasking P-TEFb complex. Curr. Opin. Cell Biol. 2008, 20, 334–340. [Google Scholar] [CrossRef]
- Shuman, S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 66, 1–40. [Google Scholar] [CrossRef]
- Cho, E.J.; Takagi, T.; Moore, C.R.; Buratowski, S. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes. Dev. 1997, 11, 3319–3326. [Google Scholar] [CrossRef]
- Wen, Y.; Shatkin, A.J. Transcription elongation factor hSPT5 stimulates mRNA capping. Genes. Dev. 1999, 13, 1774–1779. [Google Scholar] [CrossRef]
- Das, R.; Yu, J.; Zhang, Z.; Gygi, M.P.; Krainer, A.R.; Gygi, S.P.; Reed, R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 2007, 26, 867–881. [Google Scholar] [CrossRef]
- Mortillaro, M.J.; Blencowe, B.J.; Wei, X.; Nakayasu, H.; Du, L.; Warren, S.L.; Sharp, P.A.; Berezney, R. A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc. Natl. Acad. Sci. USA 1996, 93, 8253–8257. [Google Scholar]
- Yuryev, A.; Patturajan, M.; Litingtung, Y.; Joshi, R.V.; Gentile, C.; Gebara, M.; Corden, J.L. The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 6975–6980. [Google Scholar]
- Barboric, M.; Lenasi, T.; Chen, H.; Johansen, E.B.; Guo, S.; Peterlin, B.M. 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc. Natl. Acad. Sci. USA 2009, 106, 7798–7803. [Google Scholar]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Herrmann, C.H.; Gold, M.O.; Rice, A.P. Viral transactivators specifically target distinct cellular protein kinases that phosphorylate the RNA polymerase II C-terminal domain. Nucleic Acids Res. 1996, 24, 501–508. [Google Scholar] [CrossRef]
- Herrmann, C.H.; Rice, A.P. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: Candidate for a Tat cofactor. J. Virol. 1995, 69, 1612–1620. [Google Scholar]
- Marshall, N.F.; Price, D.H. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol. Cell Biol. 1992, 12, 2078–2090. [Google Scholar]
- Marshall, N.F.; Peng, J.; Xie, Z.; Price, D.H. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 1996, 271, 27176–27183. [Google Scholar]
- Yang, X.; Gold, M.O.; Tang, D.N.; Lewis, D.E.; Aguilar-Cordova, E.; Rice, A.P.; Herrmann, C.H. TAK, an HIV Tat-associated kinase, is a member of the cyclin-dependent family of protein kinases and is induced by activation of peripheral blood lymphocytes and differentiation of promonocytic cell lines. Proc. Natl. Acad. Sci. USA 1997, 94, 12331–12336. [Google Scholar]
- Zhu, Y.; Pe’ery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Garber, M.E.; Wei, P.; KewalRamani, V.N.; Mayall, T.P.; Herrmann, C.H.; Rice, A.P.; Littman, D.R.; Jones, K.A. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998, 12, 3512–3527. [Google Scholar] [CrossRef]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998, 17, 7056–7065. [Google Scholar] [CrossRef]
- Kwak, Y.T.; Ivanov, D.; Guo, J.; Nee, E.; Gaynor, R.B. Role of the human and murine cyclin T proteins in regulating HIV-1 tat-activation. J. Mol. Biol. 1999, 288, 57–69. [Google Scholar] [CrossRef]
- Albrecht, T.R.; Lund, L.H.; Garcia-Blanco, M.A. Canine cyclin T1 rescues equine infectious anemia virus tat trans-activation in human cells. Virology 2000, 268, 7–11. [Google Scholar] [CrossRef]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Highly divergent lentiviral Tat proteins activate viral gene expression by a common mechanism. Mol. Cell Biol. 1999, 19, 4592–4599. [Google Scholar]
- Korin, Y.D.; Zack, J.A. Nonproductive human immunodeficiency virus type 1 infection in nucleoside-treated G0 lymphocytes. J. Virol. 1999, 73, 6526–6532. [Google Scholar]
- Bukrinsky, M.I.; Sharova, N.; Dempsey, M.P.; Stanwick, T.L.; Bukrinskaya, A.G.; Haggerty, S.; Stevenson, M. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. USA 1992, 89, 6580–6584. [Google Scholar]
- Lassen, K.G.; Bailey, J.R.; Siliciano, R.F. Analysis of human immunodeficiency virus type 1 transcriptional elongation in resting CD4+ T cells in vivo. J. Virol. 2004, 78, 9105–9114. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Tong-Starksen, S.E.; Luciw, P.A.; Peterlin, B.M. Human immunodeficiency virus long terminal repeat responds to T-cell activation signals. Proc. Natl. Acad. Sci. USA 1987, 84, 6845–6849. [Google Scholar] [CrossRef]
- Lassen, K.G.; Ramyar, K.X.; Bailey, J.R.; Zhou, Y.; Siliciano, R.F. Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathog. 2006, 2, e68. [Google Scholar] [CrossRef]
- Tamaru, H. Confining euchromatin/heterochromatin territory: Jumonji crosses the line. Genes. Dev. 2010, 24, 1465–1478. [Google Scholar] [CrossRef]
- Herrmann, C.H.; Carroll, R.G.; Wei, P.; Jones, K.A.; Rice, A.P. Tat-associated kinase, TAK, activity is regulated by distinct mechanisms in peripheral blood lymphocytes and promonocytic cell lines. J. Virol. 1998, 72, 9881–9888. [Google Scholar]
- Liu, H.; Rice, A.P. Genomic organization and characterization of promoter function of the human CDK9 gene. Gene 2000, 252, 51–59. [Google Scholar] [CrossRef]
- Shore, S.M.; Byers, S.A.; Maury, W.; Price, D.H. Identification of a novel isoform of Cdk9. Gene 2003, 307, 175–182. [Google Scholar] [CrossRef]
- Herrmann, C.H.; Mancini, M.A. The Cdk9 and cyclin T subunits of TAK/P-TEFb localize to splicing factor-rich nuclear speckle regions. J. Cell Sci. 2001, 114, 1491–1503. [Google Scholar]
- Pendergrast, P.S.; Wang, C.; Hernandez, N.; Huang, S. FBI-1 can stimulate HIV-1 Tat activity and is targeted to a novel subnuclear domain that includes the Tat-P-TEFb-containing nuclear speckles. Mol. Biol. Cell 2002, 13, 915–929. [Google Scholar] [CrossRef]
- Liu, H.; Herrmann, C.H. Differential localization and expression of the Cdk9 42k and 55k isoforms. J. Cell Physiol. 2005, 203, 251–260. [Google Scholar] [CrossRef]
- Ghose, R.; Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J. Virol. 2001, 75, 11336–11343. [Google Scholar] [CrossRef]
- Chen, R.; Yang, Z.; Zhou, Q. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J. Biol. Chem. 2004, 279, 4153–4160. [Google Scholar]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.E.; Knapp, S.; Johnson, L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Barboric, M.; Zhang, F.; Besenicar, M.; Plemenitas, A.; Peterlin, B.M. Ubiquitylation of Cdk9 by Skp2 facilitates optimal Tat transactivation. J. Virol. 2005, 79, 11135–11141. [Google Scholar] [CrossRef]
- Sabo, A.; Lusic, M.; Cereseto, A.; Giacca, M. Acetylation of conserved lysines in the catalytic core of cyclin-dependent kinase 9 inhibits kinase activity and regulates transcription. Mol. Cell Biol. 2008, 28, 2201–2212. [Google Scholar] [CrossRef]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Pavletich, N.P. Mechanisms of cyclin-dependent kinase regulation: Structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J. Mol. Biol. 1999, 287, 821–828. [Google Scholar] [CrossRef]
- Li, Q.; Price, J.P.; Byers, S.A.; Cheng, D.; Peng, J.; Price, D.H. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar]
- Wang, Y.; Dow, E.C.; Liang, Y.Y.; Ramakrishnan, R.; Liu, H.; Sung, T.L.; Lin, X.; Rice, A.P. Phosphatase PPM1A regulates phosphorylation of Thr-186 in the Cdk9 T-loop. J. Biol. Chem. 2008, 283, 33578–33584. [Google Scholar]
- Ramakrishnan, R.; Dow, E.C.; Rice, A.P. Characterization of Cdk9 T-loop phosphorylation in resting and activated CD4(+) T lymphocytes. J. Leukoc. Biol. 2009, 86, 1345–1350. [Google Scholar] [CrossRef]
- Dong, C.; Kwas, C.; Wu, L. Transcriptional restriction of human immunodeficiency virus type 1 gene expression in undifferentiated primary monocytes. J. Virol. 2009, 83, 3518–3527. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Rice, A.P. Cdk9 T-loop phosphorylation is regulated by the calcium signaling pathway. J. Cell Physiol. 2012, 227, 609–617. [Google Scholar] [CrossRef]
- Ammosova, T.; Obukhov, Y.; Kotelkin, A.; Breuer, D.; Beullens, M.; Gordeuk, V.R.; Bollen, M.; Nekhai, S. Protein phosphatase-1 activates CDK9 by dephosphorylating Ser175. PLoS One 2011, 6, e18985. [Google Scholar]
- Chen, R.; Liu, M.; Li, H.; Xue, Y.; Ramey, W.N.; He, N.; Ai, N.; Luo, H.; Zhu, Y.; Zhou, N.; Zhou, Q. PP2B and PP1{alpha} cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008, 22, 1356–1368. [Google Scholar] [CrossRef]
- Ammosova, T.; Yedavalli, V.R.; Niu, X.; Jerebtsova, M.; van Eynde, A.; Beullens, M.; Bollen, M.; Jeang, K.T.; Nekhai, S. Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV-1 transcription. J. Biol. Chem. 2011, 286, 3798–3804. [Google Scholar]
- Li, Q.; Cooper, J.J.; Altwerger, G.H.; Feldkamp, M.D.; Shea, M.A.; Price, D.H. HEXIM1 is a promiscuous double-stranded RNA-binding protein and interacts with RNAs in addition to 7SK in cultured cells. Nucleic Acids Res. 2007, 35, 2503–2512. [Google Scholar]
- Chen, H.; Li, C.; Huang, J.; Cung, T.; Seiss, K.; Beamon, J.; Carrington, M.F.; Porter, L.C.; Burke, P.S.; Yang, Y.; et al. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J. Clin. Invest. 2011, 121, 1549–1560. [Google Scholar]
- Ammosova, T.; Berro, R.; Jerebtsova, M.; Jackson, A.; Charles, S.; Klase, Z.; Southerland, W.; Gordeuk, V.R.; Kashanchi, F.; Nekhai, S. Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology 2006, 3, 78. [Google Scholar] [CrossRef]
- Debebe, Z.; Ammosova, T.; Breuer, D.; Lovejoy, D.B.; Kalinowski, D.S.; Kumar, K.; Jerebtsova, M.; Ray, P.; Kashanchi, F.; Gordeuk, V.R.; et al. Iron chelators of the di-2-pyridylketone thiosemicarbazone and 2-benzoylpyridine thiosemicarbazone series inhibit HIV-1 transcription: Identification of novel cellular targets--iron, cyclin-dependent kinase (CDK) 2, and CDK9. Mol. Pharmacol. 2011, 79, 185–196. [Google Scholar] [CrossRef]
- Stewart, Z.A.; Leach, S.D.; Pietenpol, J.A. p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol. Cell Biol. 1999, 19, 205–215. [Google Scholar]
- Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J. Virol. 2002, 76, 10579–10587. [Google Scholar] [CrossRef]
- Liou, L.Y.; Herrmann, C.H.; Rice, A.P. HIV-1 infection and regulation of Tat function in macrophages. Int. J. Biochem. Cell Biol. 2004, 36, 1767–1775. [Google Scholar] [CrossRef]
- Rechsteiner, M.; Rogers, S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar]
- Kurosu, T.; Peterlin, B.M. VP16 and ubiquitin; binding of P-TEFb via its activation domain and ubiquitin facilitates elongation of transcription of target genes. Curr. Biol. 2004, 14, 1112–1116. [Google Scholar] [CrossRef]
- Chiang, K.; Sung, T.L.; Rice, A.P. Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef]
- Sung, T.L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Nathans, R.; Chu, C.Y.; Serquina, A.K.; Lu, C.C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef]
- Hoque, M.; Shamanna, R.A.; Guan, D.; Pe’ery, T.; Mathews, M.B. HIV-1 replication and latency are regulated by translational control of cyclin T1. J. Mol. Biol. 2011, 410, 917–932. [Google Scholar] [CrossRef]
- Agbottah, E.T.; Traviss, C.; McArdle, J.; Karki, S.; St Laurent, G.C., 3rd; Kumar, A. Nuclear Factor 90(NF90) targeted to TAR RNA inhibits transcriptional activation of HIV-1. Retrovirology 2007, 4, 41. [Google Scholar] [CrossRef]
- Sung, T.L.; Rice, A.P. Effects of prostratin on Cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 2006, 3, 66. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Yu, W.; Rice, A.P. Limited redundancy in genes regulated by Cyclin T2 and Cyclin T1. BMC Res. Notes 2011, 4, 260. [Google Scholar] [CrossRef]
- Yu, W.; Ramakrishnan, R.; Wang, Y.; Chiang, K.; Sung, T.L.; Rice, A.P. Cyclin T1-dependent genes in activated CD4 T and macrophage cell lines appear enriched in HIV-1 co-factors. PLoS One 2008, 3, e3146. [Google Scholar]
- Yu, W.; Wang, Y.; Shaw, C.A.; Qin, X.F.; Rice, A.P. Induction of the HIV-1 Tat co-factor cyclin T1 during monocyte differentiation is required for the regulated expression of a large portion of cellular mRNAs. Retrovirology 2006, 3, 32. [Google Scholar] [CrossRef]
- Kohoutek, J.; Li, Q.; Blazek, D.; Luo, Z.; Jiang, H.; Peterlin, B.M. Cyclin T2 is essential for mouse embryogenesis. Mol. Cell Biol. 2009, 29, 3280–3285. [Google Scholar] [CrossRef]
- de Luca, A.; Tosolini, A.; Russo, P.; Severino, A.; Baldi, A.; de Luca, L.; Cavallotti, I.; Baldi, F.; Giordano, A.; Testa, J.R.; Paggi, M.G. Cyclin T2a gene maps on human chromosome 2q21. J. Histochem. Cytochem. 2001, 49, 693–698. [Google Scholar] [CrossRef]
- de Luca, A.; de Falco, M.; Baldi, A.; Paggi, M.G. Cyclin T: Three forms for different roles in physiological and pathological functions. J. Cell Physiol. 2003, 194, 101–107. [Google Scholar] [CrossRef]
- Choo, S.; Schroeder, S.; Ott, M. CYCLINg through transcription: Posttranslational modifications of P-TEFb regulate transcription elongation. Cell Cycle 2010, 9, 1697–1705. [Google Scholar] [CrossRef]
- Michels, A.A.; Nguyen, V.T.; Fraldi, A.; Labas, V.; Edwards, M.; Bonnet, F.; Lania, L.; Bensaude, O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell Biol. 2003, 23, 4859–4869. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef]
- Krueger, B.J.; Varzavand, K.; Cooper, J.J.; Price, D.H. The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS One 2010, 5, e12335. [Google Scholar]
- Muniz, L.; Egloff, S.; Ughy, B.; Jady, B.E.; Kiss, T. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010, 6, e1001152. [Google Scholar] [CrossRef]
- Markert, A.; Grimm, M.; Martinez, J.; Wiesner, J.; Meyerhans, A.; Meyuhas, O.; Sickmann, A.; Fischer, U. The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep. 2008, 9, 569–575. [Google Scholar] [CrossRef]
- He, N.; Jahchan, N.S.; Hong, E.; Li, Q.; Bayfield, M.A.; Maraia, R.J.; Luo, K.; Zhou, Q. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol. Cell 2008, 29, 588–599. [Google Scholar] [CrossRef]
- Xue, Y.; Yang, Z.; Chen, R.; Zhou, Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2010, 38, 360–369. [Google Scholar] [CrossRef]
- Diribarne, G.; Bensaude, O. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol. 2009, 6, 122–128. [Google Scholar] [CrossRef]
- Blazek, D.; Barboric, M.; Kohoutek, J.; Oven, I.; Peterlin, B.M. Oligomerization of HEXIM1 via 7SK snRNA and coiled-coil region directs the inhibition of P-TEFb. Nucleic Acids Res. 2005, 33, 7000–7010. [Google Scholar] [CrossRef]
- Zhou, M.; Huang, K.; Jung, K.J.; Cho, W.K.; Klase, Z.; Kashanchi, F.; Pise-Masison, C.A.; Brady, J.N. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol. 2009, 83, 1036–1044. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.R.; Nazarian, A.A.; Li, C.C.; Gore, S.L.; Sridharan, R.; Imbalzano, A.N.; Smale, S.T. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes. Dev. 2006, 20, 282–296. [Google Scholar] [CrossRef]
- O’Malley, B.W.; Qin, J.; Lanz, R.B. Cracking the coregulator codes. Curr. Opin. Cell Biol. 2008, 20, 310–315. [Google Scholar] [CrossRef]
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the human endogenous coregulator complexome. Cell 2011, 145, 787–799. [Google Scholar] [CrossRef]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef]
- Strain, M.C.; Little, S.J.; Daar, E.S.; Havlir, D.V.; Gunthard, H.F.; Lam, R.Y.; Daly, O.A.; Nguyen, J.; Ignacio, C.C.; Spina, C.A.; et al. Effect of treatment, during primary infection, on establishment and clearance of cellular reservoirs of HIV-1. J. Infect. Dis. 2005, 191, 1410–1418. [Google Scholar] [CrossRef]
- Han, Y.; Lassen, K.; Monie, D.; Sedaghat, A.R.; Shimoji, S.; Liu, X.; Pierson, T.C.; Margolick, J.B.; Siliciano, R.F.; Siliciano, J.D. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 2004, 78, 6122–6133. [Google Scholar]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar]
- Vatakis, D.N.; Kim, S.; Kim, N.; Chow, S.A.; Zack, J.A. Human immunodeficiency virus integration efficiency and site selection in quiescent CD4+ T cells. J. Virol. 2009, 83, 6222–6233. [Google Scholar] [CrossRef]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef]
- Weinberger, L.S.; Dar, R.D.; Simpson, M.L. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat. Genet. 2008, 40, 466–470. [Google Scholar] [CrossRef]
- Yukl, S.; Pillai, S.; Li, P.; Chang, K.; Pasutti, W.; Ahlgren, C.; Havlir, D.; Strain, M.; Gunthard, H.; Richman, D.; et al. Latently-infected CD4+ T cells are enriched for HIV-1 Tat variants with impaired transactivation activity. Virology 2009, 387, 98–108. [Google Scholar] [CrossRef]
- Choudhary, S.K.; Archin, N.M.; Margolis, D.M. Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells. J. Infect. Dis. 2008, 197, 1162–1170. [Google Scholar] [CrossRef]
- Chen, R.; Liu, M.; Li, H.; Xue, Y.; Ramey, W.N.; He, N.; Ai, N.; Luo, H.; Zhu, Y.; Zhou, N.; et al. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008, 22, 1356–1368. [Google Scholar] [CrossRef]
- Contreras, X.; Barboric, M.; Lenasi, T.; Peterlin, B.M. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007, 3, 1459–1469. [Google Scholar]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar]
- Mbonye, U.; Karn, J. Control of HIV latency by epigenetic and non-epigenetic mechanisms. Curr. HIV Res. 2011, 9, 554–567. [Google Scholar] [CrossRef]
- Sahu, G.K.; Lee, K.; Ji, J.; Braciale, V.; Baron, S.; Cloyd, M.W. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology 2006, 355, 127–137. [Google Scholar] [CrossRef]
- Lassen, K.G.; Hebbeler, A.M.; Bhattacharyya, D.; Lobritz, M.A.; Greene, W.C. A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One 2012, 7, e30176. [Google Scholar]
- Saleh, S.; Solomon, A.; Wightman, F.; Xhilaga, M.; Cameron, P.U.; Lewin, S.R. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: A novel model of HIV-1 latency. Blood 2007, 110, 4161–4164. [Google Scholar]
- Yang, H.C.; Xing, S.; Shan, L.; O’Connell, K.; Dinoso, J.; Shen, A.; Zhou, Y.; Shrum, C.K.; Han, Y.; Liu, J.O.; et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J. Clin. Invest. 2009, 119, 3473–3486. [Google Scholar]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef]
- McNamara, L.A.; Collins, K.L. Hematopoietic stem/precursor cells as HIV reservoirs. Curr. Opin. HIV AIDS 2011, 6, 43–48. [Google Scholar]
- Durand, C.M.; Ghiaur, G.; Siliciano, J.D.; Rabi, S.A.; Eisele, E.E.; Salgado, M.; Shan, L.; Lai, J.F.; Zhang, H.; Margolick, J.; et al. HIV-1 DNA is detected in bone marrow populations containing CD4+ T cells but is not found in purified CD34+ hematopoietic progenitor cells in most patients on antiretroviral therapy. J. Infect. Dis. 2012, 205, 1014–1018. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ramakrishnan, R.; Chiang, K.; Liu, H.; Budhiraja, S.; Donahue, H.; Rice, A.P. Making a Short Story Long: Regulation of P-TEFb and HIV-1 Transcriptional Elongation in CD4+ T Lymphocytes and Macrophages. Biology 2012, 1, 94-115. https://doi.org/10.3390/biology1010094
Ramakrishnan R, Chiang K, Liu H, Budhiraja S, Donahue H, Rice AP. Making a Short Story Long: Regulation of P-TEFb and HIV-1 Transcriptional Elongation in CD4+ T Lymphocytes and Macrophages. Biology. 2012; 1(1):94-115. https://doi.org/10.3390/biology1010094
Chicago/Turabian StyleRamakrishnan, Rajesh, Karen Chiang, Hongbing Liu, Sona Budhiraja, Hart Donahue, and Andrew P. Rice. 2012. "Making a Short Story Long: Regulation of P-TEFb and HIV-1 Transcriptional Elongation in CD4+ T Lymphocytes and Macrophages" Biology 1, no. 1: 94-115. https://doi.org/10.3390/biology1010094
APA StyleRamakrishnan, R., Chiang, K., Liu, H., Budhiraja, S., Donahue, H., & Rice, A. P. (2012). Making a Short Story Long: Regulation of P-TEFb and HIV-1 Transcriptional Elongation in CD4+ T Lymphocytes and Macrophages. Biology, 1(1), 94-115. https://doi.org/10.3390/biology1010094