Mitochondrial Impairment in Sarcopenia



Abstract
Simple Summary
Abstract
1. Introduction
2. Aging and Skeletal Muscle Quality
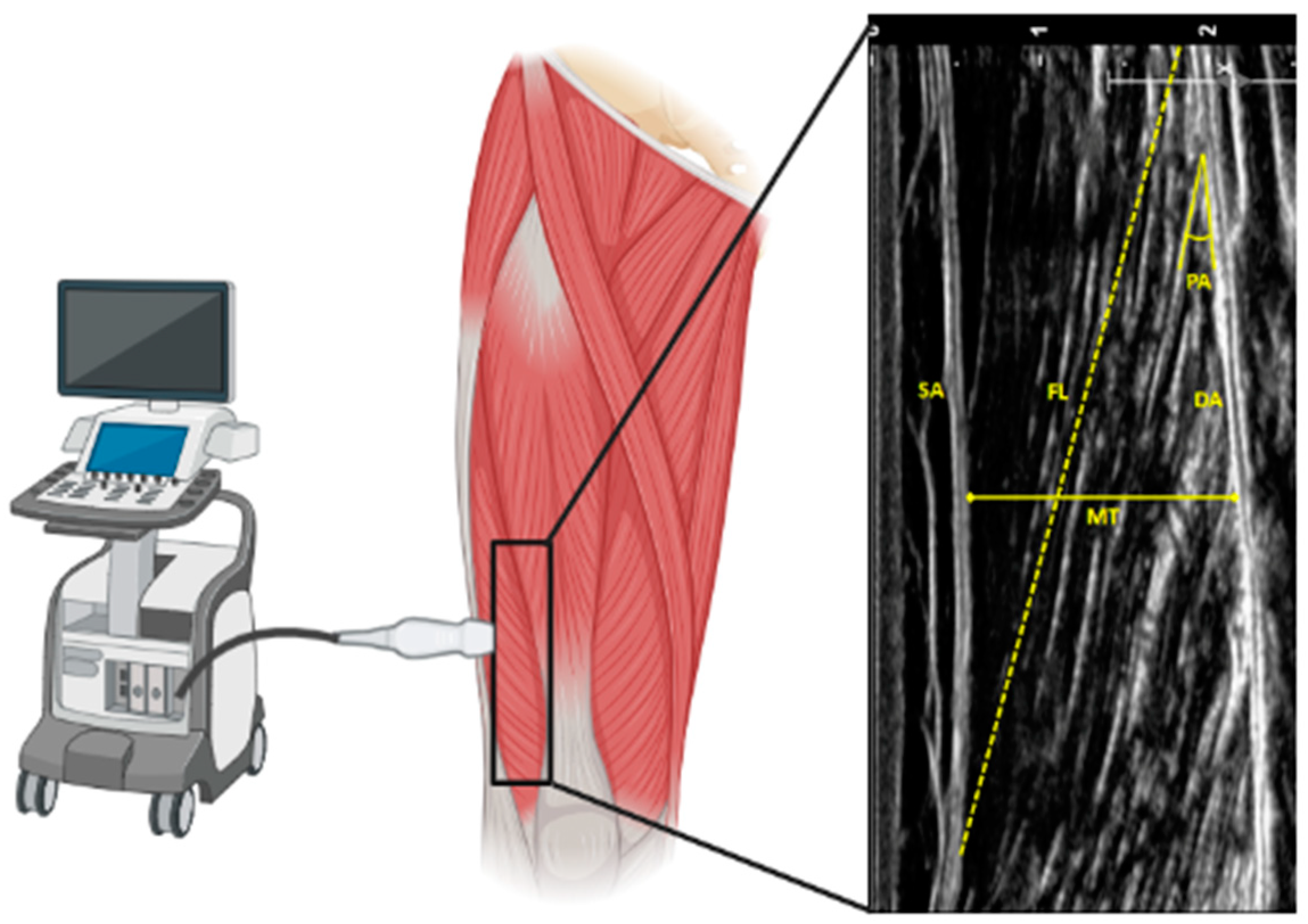
2.1. Age and Skeletal Muscle Architecture
2.2. Age and Skeletal Muscle Metabolism
3. Skeletal Muscle Mitochondria Homeostasis
4. Age-Related Mitochondrial Alterations and Sarcopenia
5. Age-Related Apoptosis and Sarcopenia
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenberg, I.H. Sarcopenia: Origins and clinical relevance. J. Nutr. 1997, 127, 990S–991S. [Google Scholar] [CrossRef]
- Pacifico, J.; Geerlings, M.A.J.; Reijnierse, E.M.; Phassouliotis, C.; Lim, W.K.; Maier, A.B. Prevalence of sarcopenia as a comorbid disease: A systematic review and meta-analysis. Exp. Gerontol. 2020, 131, 110801. [Google Scholar] [CrossRef]
- Riuzzi, F.; Sorci, G.; Arcuri, C.; Giambanco, I.; Bellezza, I.; Minelli, A.; Donato, R. Cellular and molecular mechanisms of sarcopenia: The S100B perspective. J. Cachexia Sarcopenia Muscle 2018, 9, 1255–1268. [Google Scholar] [CrossRef]
- Kwon, Y.N.; Yoon, S.S. Sarcopenia: Neurological Point of View. J. Bone Metab. 2017, 24, 83–89. [Google Scholar] [CrossRef]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Dennison, E.M.; Sayer, A.A.; Cooper, C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 340–347. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef]
- Evans, W.J. Skeletal muscle loss: Cachexia, sarcopenia, and inactivity. Am. J. Clin. Nutr. 2010, 91, 1123S–1127S. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.K.; Williams, J.; Atherton, P.; Larvin, M.; Lund, J.; Narici, M. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front. Physiol. 2012, 3, 260. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 601. [Google Scholar] [CrossRef] [PubMed]
- Landi, F.; Calvani, A.; Picca, M.; Tosato, A.; Bernabei, R.; Marzetti, E. Emerging reseach on importance of muscle mass and function. J. Gerontol. Geriatr. 2019, 67, 26–31. [Google Scholar]
- Auyeung, T.W.; Lee, S.W.; Leung, J.; Kwok, T.; Woo, J. Age-associated decline of muscle mass, grip strength and gait speed: A 4-year longitudinal study of 3018 community-dwelling older Chinese. Geriatr. Gerontol. Int. 2014, 14 (Suppl. 1), 76–84. [Google Scholar] [CrossRef] [PubMed]
- McGregor, R.A.; Cameron-Smith, D.; Poppitt, S.D. It is not just muscle mass: A review of muscle quality, composition and metabolism during ageing as determinants of muscle function and mobility in later life. Longev. Healthspan 2014, 3, 9. [Google Scholar] [CrossRef]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef]
- Martone, A.M.; Marzetti, E.; Calvani, A.; Picca, A.; Tosato, A.; Bernabei, R.; Landi, F. Assessment of sarcopenia: From clinical practice to research. J. Gerontol. Geriatr. 2019, 67, 39–45. [Google Scholar]
- Narici, M.V.; Maganaris, C.N.; Reeves, N.D.; Capodaglio, P. Effect of aging on human muscle architecture. J. Appl. Physiol. (1985) 2003, 95, 2229–2234. [Google Scholar] [CrossRef]
- Thom, J.M.; Morse, C.I.; Birch, K.M.; Narici, M.V. Influence of muscle architecture on the torque and power-velocity characteristics of young and elderly men. Eur. J. Appl. Physiol. 2007, 100, 613–619. [Google Scholar] [CrossRef]
- Lexell, J.; Taylor, C.C.; Sjostrom, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Scott, W.; Stevens, J.; Binder-Macleod, S.A. Human skeletal muscle fiber type classifications. Phys. Ther. 2001, 81, 1810–1816. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Piasecki, M.; Atherton, P.J. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res. Rev. 2018, 47, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Beavers, K.M.; Beavers, D.P.; Houston, D.K.; Harris, T.B.; Hue, T.F.; Koster, A.; Newman, A.B.; Simonsick, E.M.; Studenski, S.A.; Nicklas, B.J.; et al. Associations between body composition and gait-speed decline: Results from the Health, Aging, and Body Composition study. Am. J. Clin. Nutr. 2013, 97, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.L.; Addison, O.; Kidde, J.P.; Dibble, L.E.; Lastayo, P.C. Skeletal muscle fat infiltration: Impact of age, inactivity, and exercise. J. Nutr. Health Aging 2010, 14, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Cholok, D.; Lee, E.; Lisiecki, J.; Agarwal, S.; Loder, S.; Ranganathan, K.; Qureshi, A.T.; Davis, T.A.; Levi, B. Traumatic muscle fibrosis: From pathway to prevention. J. Trauma Acute Care Surg. 2017, 82, 174–184. [Google Scholar] [CrossRef]
- Tieland, M.; Trouwborst, I.; Clark, B.C. Skeletal muscle performance and ageing. J. Cachexia Sarcopenia Muscle 2018, 9, 3–19. [Google Scholar] [CrossRef]
- D’Antona, G.; Pellegrino, M.A.; Adami, R.; Rossi, R.; Carlizzi, C.N.; Canepari, M.; Saltin, B.; Bottinelli, R. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J. Physiol. 2003, 552, 499–511. [Google Scholar] [CrossRef]
- Kragstrup, T.W.; Kjaer, M.; Mackey, A.L. Structural, biochemical, cellular, and functional changes in skeletal muscle extracellular matrix with aging. Scand. J. Med. Sci. Sports 2011, 21, 749–757. [Google Scholar] [CrossRef]
- Westerblad, H.; Bruton, J.D.; Katz, A. Skeletal muscle: Energy metabolism, fiber types, fatigue and adaptability. Exp. Cell. Res. 2010, 316, 3093–3099. [Google Scholar] [CrossRef]
- Biolo, G.; Cederholm, T.; Muscaritoli, M. Muscle contractile and metabolic dysfunction is a common feature of sarcopenia of aging and chronic diseases: From sarcopenic obesity to cachexia. Clin. Nutr. 2014, 33, 737–748. [Google Scholar] [CrossRef]
- Gheller, B.J.; Riddle, E.S.; Lem, M.R.; Thalacker-Mercer, A.E. Understanding Age-Related Changes in Skeletal Muscle Metabolism: Differences between Females and Males. Annu. Rev. Nutr. 2016, 36, 129–156. [Google Scholar] [CrossRef]
- Croley, A.N.; Zwetsloot, K.A.; Westerkamp, L.M.; Ryan, N.A.; Pendergast, A.M.; Hickner, R.C.; Pofahl, W.E.; Gavin, T.P. Lower capillarization, VEGF protein, and VEGF mRNA response to acute exercise in the vastus lateralis muscle of aged vs. young women. J. Appl. Physiol. (1985) 2005, 99, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Coggan, A.R.; Spina, R.J.; King, D.S.; Rogers, M.A.; Brown, M.; Nemeth, P.M.; Holloszy, J.O. Skeletal muscle adaptations to endurance training in 60- to 70-yr-old men and women. J. Appl. Physiol. (1985) 1992, 72, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.N.; Sinning, W.E.; Walro, J.M.; Sieck, G.C.; Lemon, P.W. Oxidative capacity of human muscle fiber types: Effects of age and training status. J. Appl. Physiol. (1985) 1995, 78, 2033–2038. [Google Scholar] [CrossRef]
- Landers-Ramos, R.Q.; Prior, S.J. The Microvasculature and Skeletal Muscle Health in Aging. Exerc. Sport Sci. Rev. 2018, 46, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Gaster, M.; Poulsen, P.; Handberg, A.; Schroder, H.D.; Beck-Nielsen, H. Direct evidence of fiber type-dependent GLUT-4 expression in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E910–E916. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S.; Reggiani, C.; Mann, M. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef]
- Consitt, L.A.; Dudley, C.; Saxena, G. Impact of Endurance and Resistance Training on Skeletal Muscle Glucose Metabolism in Older Adults. Nutrients 2019, 11, 2636. [Google Scholar] [CrossRef]
- Tucker, M.Z.; Turcotte, L.P. Impaired fatty acid oxidation in muscle of aging rats perfused under basal conditions. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1102–E1109. [Google Scholar] [CrossRef]
- Tucker, M.Z.; Turcotte, L.P. Aging is associated with elevated muscle triglyceride content and increased insulin-stimulated fatty acid uptake. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E827–E835. [Google Scholar] [CrossRef][Green Version]
- Volpi, E.; Mittendorfer, B.; Rasmussen, B.B.; Wolfe, R.R. The response of muscle protein anabolism to combined hyperaminoacidemia and glucose-induced hyperinsulinemia is impaired in the elderly. J. Clin. Endocrinol. Metab. 2000, 85, 4481–4490. [Google Scholar] [CrossRef]
- Paddon-Jones, D.; Sheffield-Moore, M.; Zhang, X.J.; Volpi, E.; Wolf, S.E.; Aarsland, A.; Ferrando, A.A.; Wolfe, R.R. Amino acid ingestion improves muscle protein synthesis in the young and elderly. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E321–E328. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Rasmussen, B.B. Skeletal muscle protein balance and metabolism in the elderly. Curr. Aging Sci. 2011, 4, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Volpi, E.; Sheffield-Moore, M.; Rasmussen, B.B.; Wolfe, R.R. Basal muscle amino acid kinetics and protein synthesis in healthy young and older men. JAMA 2001, 286, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Rennie, M.J. Anabolic resistance: The effects of aging, sexual dimorphism, and immobilization on human muscle protein turnover. Appl. Physiol. Nutr. Metab. 2009, 34, 377–381. [Google Scholar] [CrossRef]
- Cobley, J.N.; Sakellariou, G.K.; Murray, S.; Waldron, S.; Gregson, W.; Burniston, J.G.; Morton, J.P.; Iwanejko, L.A.; Close, G.L. Lifelong endurance training attenuates age-related genotoxic stress in human skeletal muscle. Longev. Healthspan 2013, 2, 11. [Google Scholar] [CrossRef]
- Cobley, J.N.; Sakellariou, G.K.; Owens, D.J.; Murray, S.; Waldron, S.; Gregson, W.; Fraser, W.D.; Burniston, J.G.; Iwanejko, L.A.; McArdle, A.; et al. Lifelong training preserves some redox-regulated adaptive responses after an acute exercise stimulus in aged human skeletal muscle. Free Radic. Biol. Med. 2014, 70, 23–32. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Deubel, S.; Jung, T.; Ost, M.; Klaus, S.; Grune, T.; Castro, J.P. Low proteasomal activity in fast skeletal muscle fibers is not associated with increased age-related oxidative damage. Exp. Gerontol. 2019, 117, 45–52. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Cogswell, A.M.; Stevens, R.J.; Hood, D.A. Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am. J. Physiol. 1993, 264, C383–C389. [Google Scholar] [CrossRef]
- Hood, D.A. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. (1985) 2001, 90, 1137–1157. [Google Scholar] [CrossRef]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. Appl. Physiol. (1985) 2013, 114, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Joseph, A.M.; Adhihetty, P.J.; Huang, J.H.; Saleem, A.; Uguccioni, G.; Hood, D.A. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany N. Y.) 2009, 1, 818–830. [Google Scholar] [CrossRef]
- Ferreira, R.; Vitorino, R.; Alves, R.M.; Appell, H.J.; Powers, S.K.; Duarte, J.A.; Amado, F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 2010, 10, 3142–3154. [Google Scholar] [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef]
- Yi, J.; Ma, C.; Li, Y.; Weisleder, N.; Rios, E.; Ma, J.; Zhou, J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 2011, 286, 32436–32443. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.R.; Cordova, A.; Valladares, D.; Llanos, P.; Hidalgo, C.; Gherardi, G.; De, S.D.; Mammucari, C.; Rizzuto, R.; Contreras-Ferrat, A.; et al. Mitochondrial Calcium Increase Induced by RyR1 and IP3R Channel Activation After Membrane Depolarization Regulates Skeletal Muscle Metabolism. Front. Physiol. 2018, 9, 791. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Dahl, R.; Larsen, S.; Dohlmann, T.L.; Qvortrup, K.; Helge, J.W.; Dela, F.; Prats, C. Three-dimensional reconstruction of the human skeletal muscle mitochondrial network as a tool to assess mitochondrial content and structural organization. Acta Physiol. (Oxf.) 2015, 213, 145–155. [Google Scholar] [CrossRef]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial Dynamics is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef]
- Iqbal, S.; Ostojic, O.; Singh, K.; Joseph, A.M.; Hood, D.A. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 2013, 48, 963–970. [Google Scholar] [CrossRef]
- Nielsen, J.; Gejl, K.D.; Hey-Mogensen, M.; Holmberg, H.C.; Suetta, C.; Krustrup, P.; Elemans, C.P.H.; Ortenblad, N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 2017, 595, 2839–2847. [Google Scholar] [CrossRef]
- Hoppeler, H. Exercise-induced ultrastructural changes in skeletal muscle. Int. J. Sports Med. 1986, 7, 187–204. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Little, J.P.; Stokl, A.J.; Hettinga, B.P.; Akhtar, M.; Tarnopolsky, M.A. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J. Biol. Chem. 2011, 286, 10605–10617. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Gurd, B.J. Deacetylation of PGC-1alpha by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef]
- Marcinek, D.J.; Schenkman, K.A.; Ciesielski, W.A.; Lee, D.; Conley, K.E. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J. Physiol. 2005, 569, 467–473. [Google Scholar] [CrossRef]
- Masuyama, M.; Iida, R.; Takatsuka, H.; Yasuda, T.; Matsuki, T. Quantitative change in mitochondrial DNA content in various mouse tissues during aging. Biochim. Biophys. Acta 2005, 1723, 302–308. [Google Scholar] [CrossRef]
- Yeo, D.; Kang, C.; Gomez-Cabrera, M.C.; Vina, J.; Ji, L.L. Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo. Free Radic. Biol. Med. 2019, 130, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, M.; Brown, A.; Merrell, E.; DiSanto-Rose, M.; Rathmacher, J.A.; Reynolds, T.H. PKB signaling and atrogene expression in skeletal muscle of aged mice. J. Appl. Physiol. (1985) 2011, 111, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean, P.F.; Sgarioto, N.; Auger, M.J.; Vallee, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef]
- Koltai, E.; Hart, N.; Taylor, A.W.; Goto, S.; Ngo, J.K.; Davies, K.J.; Radak, Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R127–R134. [Google Scholar] [CrossRef]
- Pesce, V.; Cormio, A.; Fracasso, F.; Lezza, A.M.; Cantatore, P.; Gadaleta, M.N. Age-related changes of mitochondrial DNA content and mitochondrial genotypic and phenotypic alterations in rat hind-limb skeletal muscles. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 715–723. [Google Scholar] [CrossRef][Green Version]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Thomas, M.M.; Wright, K.J.; Hepple, R.T. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell 2011, 10, 1047–1055. [Google Scholar] [CrossRef]
- Huang, J.H.; Joseph, A.M.; Ljubicic, V.; Iqbal, S.; Hood, D.A. Effect of age on the processing and import of matrix-destined mitochondrial proteins in skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 138–146. [Google Scholar] [CrossRef]
- Joseph, A.M.; Ljubicic, V.; Adhihetty, P.J.; Hood, D.A. Biogenesis of the mitochondrial Tom40 channel in skeletal muscle from aged animals and its adaptability to chronic contractile activity. Am. J. Physiol. Cell. Physiol. 2010, 298, C1308–C1314. [Google Scholar] [CrossRef]
- Bua, E.A.; McKiernan, S.H.; Wanagat, J.; McKenzie, D.; Aiken, J.M. Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J. Appl. Physiol. (1985) 2002, 92, 2617–2624. [Google Scholar] [CrossRef]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- McKiernan, S.H.; Colman, R.; Lopez, M.; Beasley, T.M.; Weindruch, R.; Aiken, J.M. Longitudinal analysis of early stage sarcopenia in aging rhesus monkeys. Exp. Gerontol. 2009, 44, 170–176. [Google Scholar] [CrossRef]
- Boffoli, D.; Scacco, S.C.; Vergari, R.; Solarino, G.; Santacroce, G.; Papa, S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1994, 1226, 73–82. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Kent-Braun, J.A.; Ng, A.V. Skeletal muscle oxidative capacity in young and older women and men. J. Appl. Physiol. (1985) 2000, 89, 1072–1078. [Google Scholar] [CrossRef]
- Liu, D.; Sartor, M.A.; Nader, G.A.; Pistilli, E.E.; Tanton, L.; Lilly, C.; Gutmann, L.; IglayReger, H.B.; Visich, P.S.; Hoffman, E.P.; et al. Microarray analysis reveals novel features of the muscle aging process in men and women. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1035–1044. [Google Scholar] [CrossRef]
- Ghosh, S.; Lertwattanarak, R.; Lefort, N.; Molina-Carrion, M.; Joya-Galeana, J.; Bowen, B.P.; Garduno-Garcia, J.J.; Abdul-Ghani, M.; Richardson, A.; DeFronzo, R.A.; et al. Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 2011, 60, 2051–2060. [Google Scholar] [CrossRef]
- Lezza, A.M.; Pesce, V.; Cormio, A.; Fracasso, F.; Vecchiet, J.; Felzani, G.; Cantatore, P.; Gadaleta, M.N. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 in skeletal muscle from aged human subjects. FEBS Lett. 2001, 501, 74–78. [Google Scholar] [CrossRef]
- Melov, S.; Shoffner, J.M.; Kaufman, A.; Wallace, D.C. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 1995, 23, 4122–4126. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Lee, C.C.; Vandiver, A.R.; Aiken, J.M.; McKenzie, D.; Hoang, A.; Allison, D.; Liu, N.; Wanagat, J. Mitochondrial DNA deletion mutations increase exponentially with age in human skeletal muscle. Aging Clin. Exp. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Hamadeh, M.J.; Kaczor, J.J.; Raha, S.; deBeer, J.; Tarnopolsky, M.A. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE 2010, 5, e10778. [Google Scholar] [CrossRef]
- Callahan, D.M.; Bedrin, N.G.; Subramanian, M.; Berking, J.; Ades, P.A.; Toth, M.J.; Miller, M.S. Age-related structural alterations in human skeletal muscle fibers and mitochondria are sex specific: Relationship to single-fiber function. J. Appl. Physiol. (1985) 2014, 116, 1582–1592. [Google Scholar] [CrossRef]
- Joseph, A.M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef]
- Marzetti, E.; Leeuwenburgh, C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp. Gerontol. 2006, 41, 1234–1238. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Gijsen, A.P.; Saris, W.H.; Soeters, P.B.; Wagenmakers, A.J. Derangement in aerobic and anaerobic energy metabolism in skeletal muscle of critically ill and recovering rats. Biochim. Biophys. Acta 1996, 1315, 55–60. [Google Scholar] [CrossRef]
- Johnson, M.L.; Robinson, M.M.; Nair, K.S. Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 2013, 24, 247–256. [Google Scholar] [CrossRef]
- Carter, H.N.; Chen, C.C.; Hood, D.A. Mitochondria, muscle health, and exercise with advancing age. Physiology (Bethesda) 2015, 30, 208–223. [Google Scholar] [CrossRef] [PubMed]
- Beregi, E.; Regius, O.; Huttl, T.; Gobl, Z. Age-related changes in the skeletal muscle cells. Z. Gerontol. 1988, 21, 83–86. [Google Scholar] [PubMed]
- Aiken, J.; Bua, E.; Cao, Z.; Lopez, M.; Wanagat, J.; McKenzie, D.; McKiernan, S. Mitochondrial DNA deletion mutations and sarcopenia. Ann. N. Y. Acad. Sci. 2002, 959, 412–423. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, D.; Bua, E.; McKiernan, S.; Cao, Z.; Aiken, J.M. Mitochondrial DNA deletion mutations: A causal role in sarcopenia. Eur. J. Biochem. 2002, 269, 2010–2015. [Google Scholar] [CrossRef]
- Dodds, R.M.; Davies, K.; Granic, A.; Hollingsworth, K.G.; Warren, C.; Gorman, G.; Turnbull, D.M.; Sayer, A.A. Mitochondrial respiratory chain function and content are preserved in the skeletal muscle of active very old men and women. Exp. Gerontol. 2018, 113, 80–85. [Google Scholar] [CrossRef]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Grady, J.P.; Turnbull, D.M. Respiratory chain deficiency in aged spinal motor neurons. Neurobiol. Aging 2014, 35, 2230–2238. [Google Scholar] [CrossRef]
- Pollock, N.; Staunton, C.A.; Vasilaki, A.; McArdle, A.; Jackson, M.J. Denervated muscle fibers induce mitochondrial peroxide generation in neighboring innervated fibers: Role in muscle aging. Free Radic. Biol. Med. 2017, 112, 84–92. [Google Scholar] [CrossRef]
- Joseph, A.M.; Adhihetty, P.J.; Wawrzyniak, N.R.; Wohlgemuth, S.E.; Picca, A.; Kujoth, G.C.; Prolla, T.A.; Leeuwenburgh, C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 2013, 8, e69327. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Lorenzi, M.; Menghi, A.; Galli, M.; Vitiello, R.; Randisi, F.; Bernabei, R.; Landi, F.; Marzetti, E. Mitochondrial dynamics signaling is shifted toward fusion in muscles of very old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp. Gerontol. 2017, 96, 63–67. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Ichim, G.; Tait, S.W.; Passos, J.F. Depletion of mitochondria in mammalian cells through enforced mitophagy. Nat. Protoc. 2017, 12, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea, D.M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [PubMed]
- Derbre, F.; Gomez-Cabrera, M.C.; Nascimento, A.L.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Tresguerres, J.A.; Fuentes, T.; Gratas-Delamarche, A.; Monsalve, M.; Vina, J. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1alpha to exercise training. Age (Dordr.) 2012, 34, 669–679. [Google Scholar] [CrossRef]
- Tamaki, M.; Miyashita, K.; Hagiwara, A.; Wakino, S.; Inoue, H.; Fujii, K.; Fujii, C.; Endo, S.; Uto, A.; Mitsuishi, M.; et al. Ghrelin treatment improves physical decline in sarcopenia model mice through muscular enhancement and mitochondrial activation. Endocr. J. 2017, 64, S47–S51. [Google Scholar] [CrossRef]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musaro, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ’exercise mimetic’ in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef]
- Vinel, C.; Lukjanenko, L.; Batut, A.; Deleruyelle, S.; Pradere, J.P.; Le, G.S.; Dortignac, A.; Geoffre, N.; Pereira, O.; Karaz, S.; et al. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018, 24, 1360–1371. [Google Scholar] [CrossRef]
- Kim, C.; Hwang, J.K. The 5,7-Dimethoxyflavone Suppresses Sarcopenia by Regulating Protein Turnover and Mitochondria Biogenesis-Related Pathways. Nutrients 2020, 12, 1079. [Google Scholar] [CrossRef]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; Lastayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, C422–C430. [Google Scholar] [CrossRef]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Beli, R.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Bucci, C.; Guerra, F.; Marzetti, E. Older Adults with Physical Frailty and Sarcopenia Show Increased Levels of Circulating Small Extracellular Vesicles with a Specific Mitochondrial Signature. Cells 2020, 9, 973. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Primeau, A.J.; Adhihetty, P.J.; Hood, D.A. Apoptosis in heart and skeletal muscle. Can. J. Appl. Physiol. 2002, 27, 349–395. [Google Scholar] [CrossRef]
- Mayer, B.; Oberbauer, R. Mitochondrial regulation of apoptosis. News Physiol. Sci. 2003, 18, 89–94. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef]
- Cande, C.; Vahsen, N.; Garrido, C.; Kroemer, G. Apoptosis-inducing factor (AIF): Caspase-independent after all. Cell Death Differ. 2004, 11, 591–595. [Google Scholar] [CrossRef]
- Dirks, A.; Leeuwenburgh, C. Apoptosis in skeletal muscle with aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R519–R527. [Google Scholar] [CrossRef]
- Phillips, T.; Leeuwenburgh, C. Muscle fiber specific apoptosis and TNF-alpha signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 2005, 19, 668–670. [Google Scholar] [CrossRef]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef]
- Song, W.; Kwak, H.B.; Lawler, J.M. Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxid. Redox Signal. 2006, 8, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Adhihetty, P.J.; Ljubicic, V.; Hood, D.A. Effect of chronic contractile activity on SS and IMF mitochondrial apoptotic susceptibility in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E748–E755. [Google Scholar] [CrossRef]
- Salucci, S.; Battistelli, M.; Baldassarri, V.; Burini, D.; Falcieri, E.; Burattini, S. Melatonin prevents mitochondrial dysfunctions and death in differentiated skeletal muscle cells. Microsc. Res. Tech. 2017, 80, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Sayed, R.K.A.; Fernandez-Ortiz, M.; Diaz-Casado, M.E.; Rusanova, I.; Rahim, I.; Escames, G.; Lopez, L.C.; Mokhtar, D.M.; Acuna-Castroviejo, D. The Protective Effect of Melatonin against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1330–1338. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Species | Age Groups Studied | Relevant Finding in Old Skeletal Muscle | Reference |
|---|---|---|---|
| C57Bl/6 Mice | 7 vs. 30 months old | ↓ mitochondrial coupling | [80] |
| C57Bl/6 Mice | 2–15 months old | ↑ TFAM and mtDNA content | [81] |
| C57Bl/6 Mice | 2 vs. 24 months old | ↑ mitophagy ↑ mitochondrial fission | [82] |
| C57Bl/6 Mice | 11–13 vs. 25–27 months old | ↓ autophagy | [83] |
| C5l/6 Mice | 3 vs. 18 months old | ↓ mitochondrial content ↓ mitochondrial biogenesis ↑ mitophagy flux | [84] |
| Mice | 8–12 vs. 88–96 weeks old | ↑ mitochondrial fusion index | [85] |
| Wistar Rats | 3 vs. 26 months old | ↓ mitochondrial mass ↓ PGC-1α protein ↑ Fis1 and Mfs1 proteins | [86] |
| Wistar Rats | 3–28 months old | ↑ mtDNA deletions ↓ respiratory enzymes | [87] |
| Fischer 344 Brown Norway Rats | 5 vs. 35 months old | ↓ mitochondrial size ↑ mitochondrial fission proteins | [67] |
| Fischer 344 Brown Norway Rats | 6 vs. 24 months old | ↓ autophagy | [88] |
| Fischer 344 Brown Norway Rats | 6 vs. 36 months old | ↓ mitochondrial content ↑ mitochondrial ROS production ↑ cytochrome c and endonuclease G release | [56] |
| Fischer 344 Brown Norway Rats | 8–10 vs. 35–36 months old | ↓ OXPHOS proteins, CS activity, state III respiration, mPTP function ↑ free radical leak | [89] |
| Fischer 344 Brown Norway Rats | 6 vs. 35–38 months old | =mitochondrial protein import machinery | [90] |
| Fischer 344 Brown Norway Rats | 6 vs. 36 months old | ↑ protein assembly =protein import | [91] |
| Fischer 344 Brown Norway Rats | 5, 18, 36 months old | ↑ ETC abnormalities | [92] |
| Fischer 344 Brown Norway Rats | 5–6 vs. 35–36 months old | ↑ mitophagy flux | [93] |
| Rhesus Monkeys | 6, 9, 12 years old | ↑ enzyme abnormalities ↑ mtDNA deletion mutations | [94] |
| Humans | 17–91 years old | ↓ respiratory activity of complex I, II, and IV | [95] |
| Humans | 18–89 years old | ↓ mtDNA, mRNA, and mitochondrial proteins ↓ mitochondrial ATP production | [96] |
| Humans | 29–80 years old | ↓ oxidative capacity | [97] |
| Humans | 22–75 years old | ↓ mitochondrial transcriptome | [98] |
| Humans | 25–72 years old | ↓ ETC proteins ↓ ROS production | [99] |
| Humans | 21–88 years old | ↑ TFAM mRNA and protein | [100] |
| Humans | 20–71 years old | ↑ mtDNA rearrangements | [101] |
| Humans | 49–93 years old | ↑ mtDNA deletion mutations | [102] |
| Humans | 20–80 years old | ↑ mtDNA deletions | [103] |
| Humans | 20–75 years old | ↓ mitochondrial enzymes activity ↓ mitochondrial biogenesis | [104] |
| Humans | 21–75 years old | ↓ IMF mitochondrial size | [105] |
| Humans | 22–82 years old | ↓ mitochondrial respiration ↓ PGC-1α, COX I, and OPA proteins ↑ mitochondrial protein import machinery | [106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellanti, F.; Lo Buglio, A.; Vendemiale, G. Mitochondrial Impairment in Sarcopenia. Biology 2021, 10, 31. https://doi.org/10.3390/biology10010031
Bellanti F, Lo Buglio A, Vendemiale G. Mitochondrial Impairment in Sarcopenia. Biology. 2021; 10(1):31. https://doi.org/10.3390/biology10010031
Chicago/Turabian StyleBellanti, Francesco, Aurelio Lo Buglio, and Gianluigi Vendemiale. 2021. "Mitochondrial Impairment in Sarcopenia" Biology 10, no. 1: 31. https://doi.org/10.3390/biology10010031
APA StyleBellanti, F., Lo Buglio, A., & Vendemiale, G. (2021). Mitochondrial Impairment in Sarcopenia. Biology, 10(1), 31. https://doi.org/10.3390/biology10010031