Mitochondrial Impairment in Sarcopenia



Abstract
:Simple Summary
Abstract
1. Introduction
2. Aging and Skeletal Muscle Quality
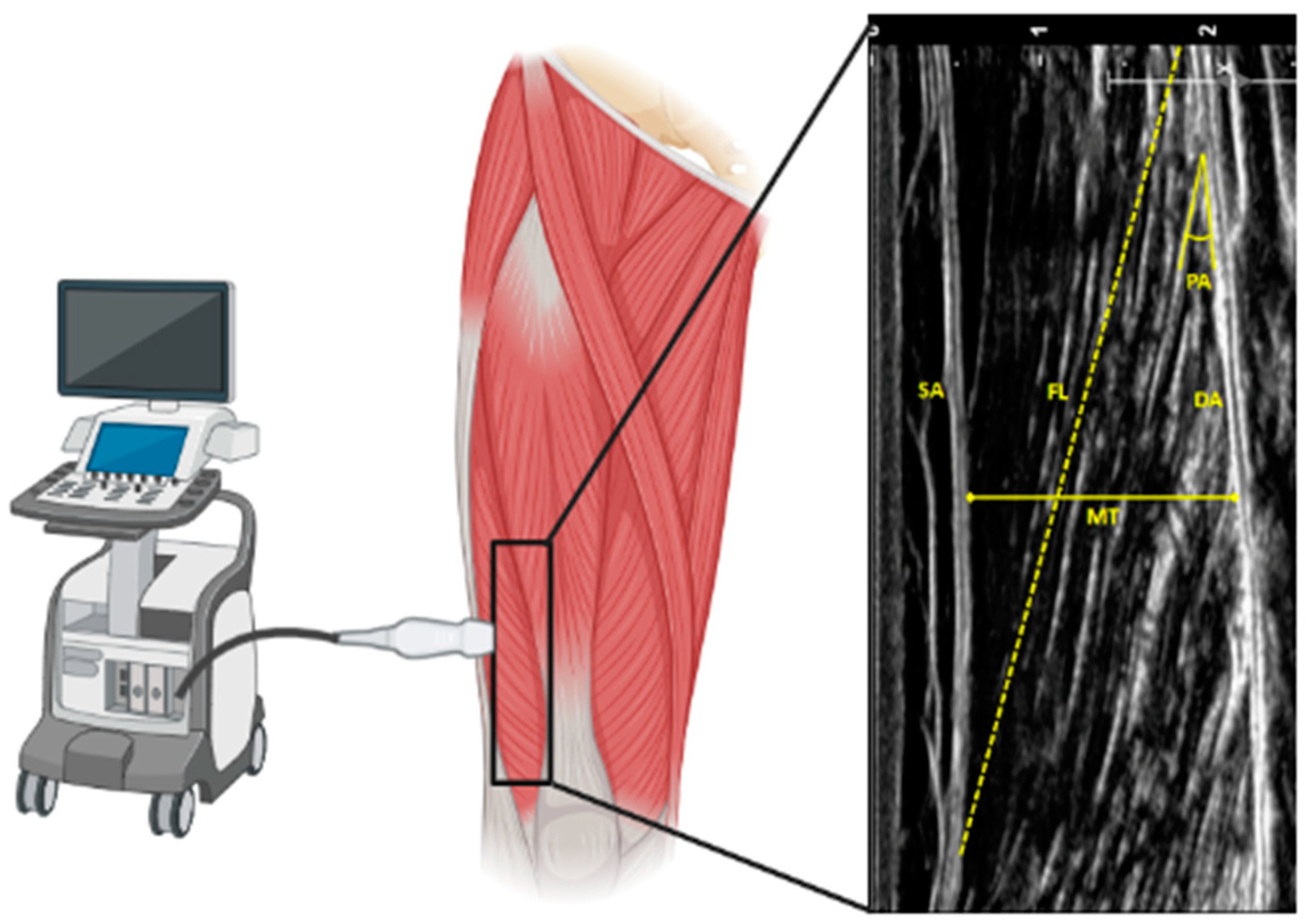
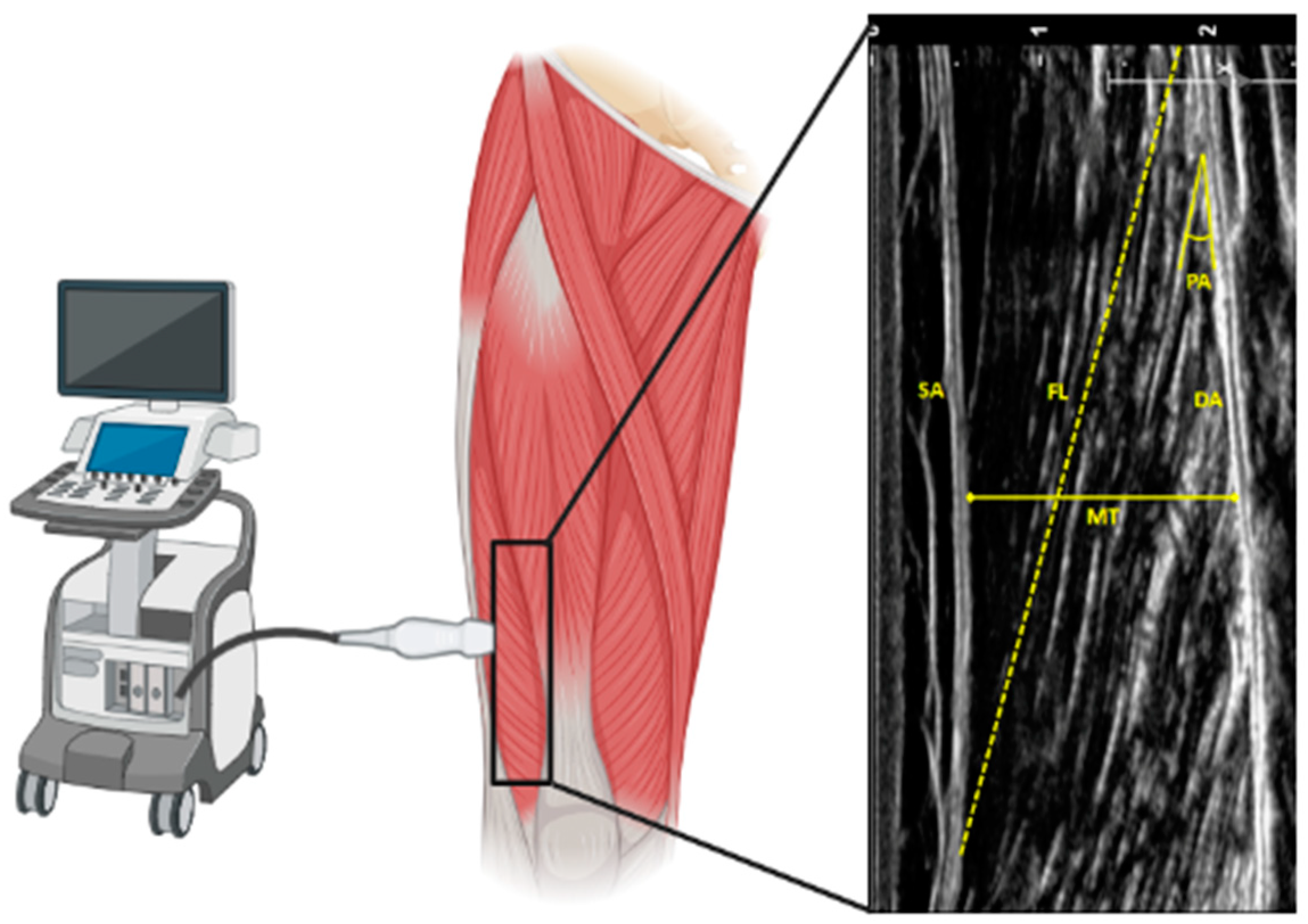
2.1. Age and Skeletal Muscle Architecture
2.2. Age and Skeletal Muscle Metabolism
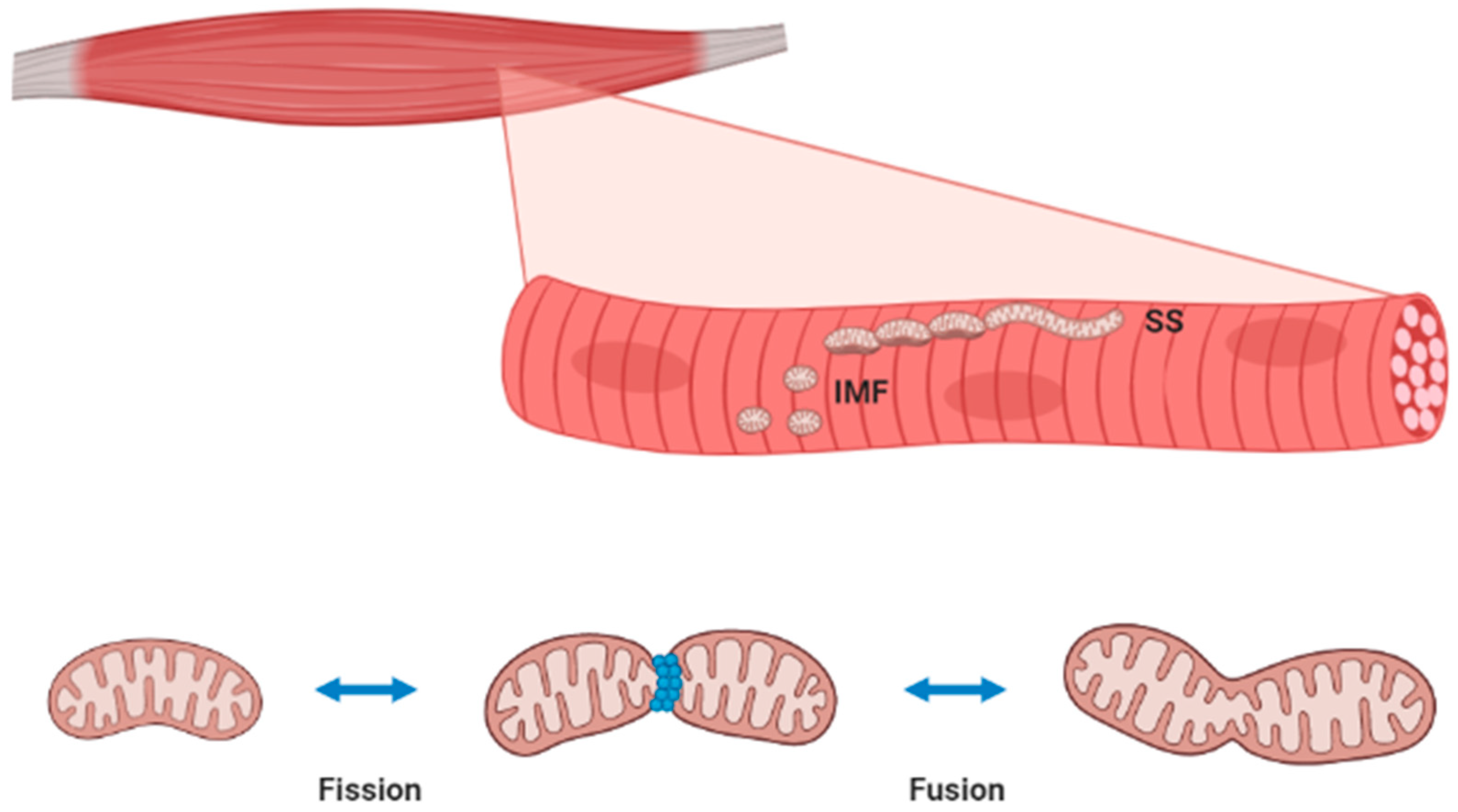
3. Skeletal Muscle Mitochondria Homeostasis
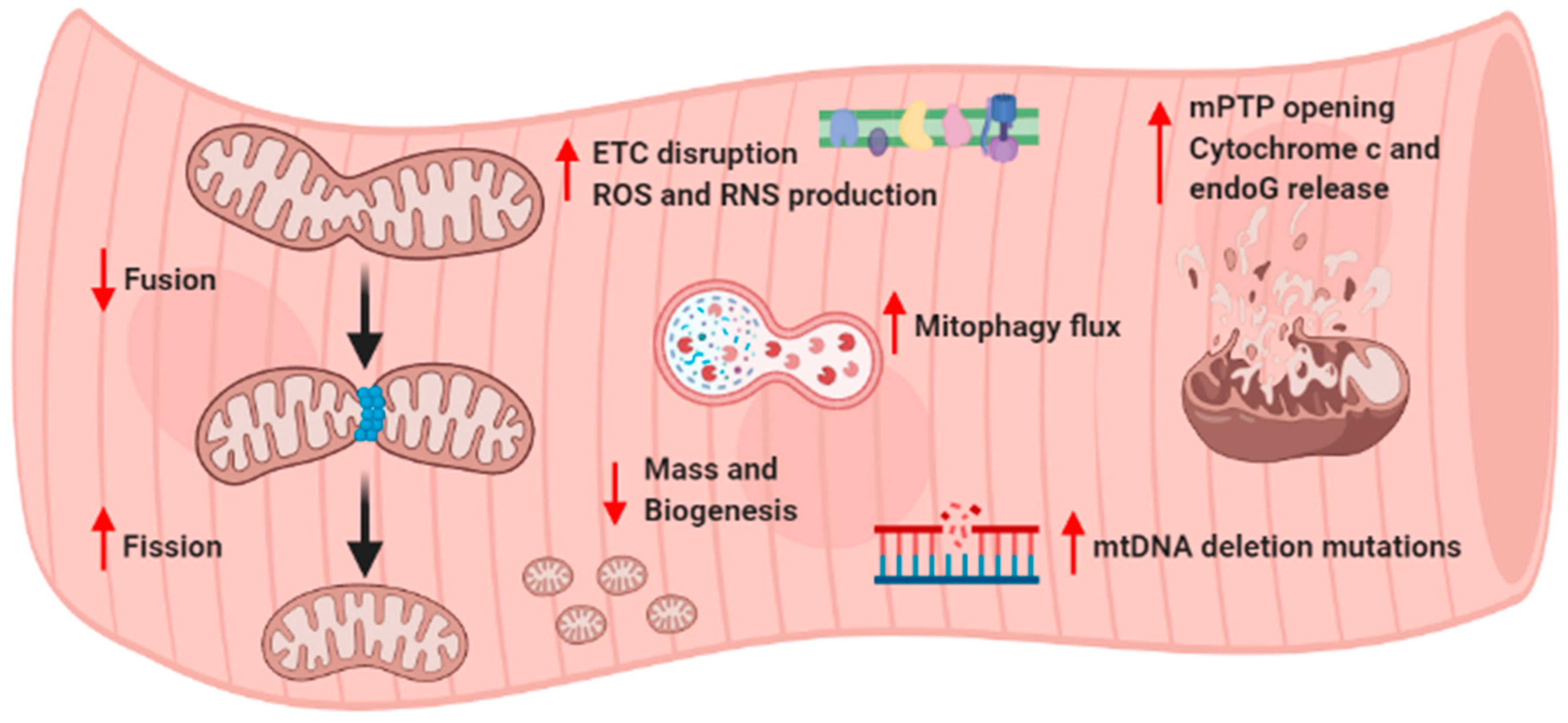
4. Age-Related Mitochondrial Alterations and Sarcopenia
5. Age-Related Apoptosis and Sarcopenia
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenberg, I.H. Sarcopenia: Origins and clinical relevance. J. Nutr. 1997, 127, 990S–991S. [Google Scholar] [CrossRef] [Green Version]
- Pacifico, J.; Geerlings, M.A.J.; Reijnierse, E.M.; Phassouliotis, C.; Lim, W.K.; Maier, A.B. Prevalence of sarcopenia as a comorbid disease: A systematic review and meta-analysis. Exp. Gerontol. 2020, 131, 110801. [Google Scholar] [CrossRef]
- Riuzzi, F.; Sorci, G.; Arcuri, C.; Giambanco, I.; Bellezza, I.; Minelli, A.; Donato, R. Cellular and molecular mechanisms of sarcopenia: The S100B perspective. J. Cachexia Sarcopenia Muscle 2018, 9, 1255–1268. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.N.; Yoon, S.S. Sarcopenia: Neurological Point of View. J. Bone Metab. 2017, 24, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Dennison, E.M.; Sayer, A.A.; Cooper, C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef]
- Evans, W.J. Skeletal muscle loss: Cachexia, sarcopenia, and inactivity. Am. J. Clin. Nutr. 2010, 91, 1123S–1127S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, W.K.; Williams, J.; Atherton, P.; Larvin, M.; Lund, J.; Narici, M. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front. Physiol. 2012, 3, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, F.; Calvani, A.; Picca, M.; Tosato, A.; Bernabei, R.; Marzetti, E. Emerging reseach on importance of muscle mass and function. J. Gerontol. Geriatr. 2019, 67, 26–31. [Google Scholar]
- Auyeung, T.W.; Lee, S.W.; Leung, J.; Kwok, T.; Woo, J. Age-associated decline of muscle mass, grip strength and gait speed: A 4-year longitudinal study of 3018 community-dwelling older Chinese. Geriatr. Gerontol. Int. 2014, 14 (Suppl. 1), 76–84. [Google Scholar] [CrossRef] [PubMed]
- McGregor, R.A.; Cameron-Smith, D.; Poppitt, S.D. It is not just muscle mass: A review of muscle quality, composition and metabolism during ageing as determinants of muscle function and mobility in later life. Longev. Healthspan 2014, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef] [Green Version]
- Martone, A.M.; Marzetti, E.; Calvani, A.; Picca, A.; Tosato, A.; Bernabei, R.; Landi, F. Assessment of sarcopenia: From clinical practice to research. J. Gerontol. Geriatr. 2019, 67, 39–45. [Google Scholar]
- Narici, M.V.; Maganaris, C.N.; Reeves, N.D.; Capodaglio, P. Effect of aging on human muscle architecture. J. Appl. Physiol. (1985) 2003, 95, 2229–2234. [Google Scholar] [CrossRef] [Green Version]
- Thom, J.M.; Morse, C.I.; Birch, K.M.; Narici, M.V. Influence of muscle architecture on the torque and power-velocity characteristics of young and elderly men. Eur. J. Appl. Physiol. 2007, 100, 613–619. [Google Scholar] [CrossRef]
- Lexell, J.; Taylor, C.C.; Sjostrom, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Scott, W.; Stevens, J.; Binder-Macleod, S.A. Human skeletal muscle fiber type classifications. Phys. Ther. 2001, 81, 1810–1816. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Piasecki, M.; Atherton, P.J. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res. Rev. 2018, 47, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Beavers, K.M.; Beavers, D.P.; Houston, D.K.; Harris, T.B.; Hue, T.F.; Koster, A.; Newman, A.B.; Simonsick, E.M.; Studenski, S.A.; Nicklas, B.J.; et al. Associations between body composition and gait-speed decline: Results from the Health, Aging, and Body Composition study. Am. J. Clin. Nutr. 2013, 97, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, R.L.; Addison, O.; Kidde, J.P.; Dibble, L.E.; Lastayo, P.C. Skeletal muscle fat infiltration: Impact of age, inactivity, and exercise. J. Nutr. Health Aging 2010, 14, 362–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholok, D.; Lee, E.; Lisiecki, J.; Agarwal, S.; Loder, S.; Ranganathan, K.; Qureshi, A.T.; Davis, T.A.; Levi, B. Traumatic muscle fibrosis: From pathway to prevention. J. Trauma Acute Care Surg. 2017, 82, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Tieland, M.; Trouwborst, I.; Clark, B.C. Skeletal muscle performance and ageing. J. Cachexia Sarcopenia Muscle 2018, 9, 3–19. [Google Scholar] [CrossRef]
- D’Antona, G.; Pellegrino, M.A.; Adami, R.; Rossi, R.; Carlizzi, C.N.; Canepari, M.; Saltin, B.; Bottinelli, R. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J. Physiol. 2003, 552, 499–511. [Google Scholar] [CrossRef]
- Kragstrup, T.W.; Kjaer, M.; Mackey, A.L. Structural, biochemical, cellular, and functional changes in skeletal muscle extracellular matrix with aging. Scand. J. Med. Sci. Sports 2011, 21, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Westerblad, H.; Bruton, J.D.; Katz, A. Skeletal muscle: Energy metabolism, fiber types, fatigue and adaptability. Exp. Cell. Res. 2010, 316, 3093–3099. [Google Scholar] [CrossRef]
- Biolo, G.; Cederholm, T.; Muscaritoli, M. Muscle contractile and metabolic dysfunction is a common feature of sarcopenia of aging and chronic diseases: From sarcopenic obesity to cachexia. Clin. Nutr. 2014, 33, 737–748. [Google Scholar] [CrossRef]
- Gheller, B.J.; Riddle, E.S.; Lem, M.R.; Thalacker-Mercer, A.E. Understanding Age-Related Changes in Skeletal Muscle Metabolism: Differences between Females and Males. Annu. Rev. Nutr. 2016, 36, 129–156. [Google Scholar] [CrossRef]
- Croley, A.N.; Zwetsloot, K.A.; Westerkamp, L.M.; Ryan, N.A.; Pendergast, A.M.; Hickner, R.C.; Pofahl, W.E.; Gavin, T.P. Lower capillarization, VEGF protein, and VEGF mRNA response to acute exercise in the vastus lateralis muscle of aged vs. young women. J. Appl. Physiol. (1985) 2005, 99, 1872–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coggan, A.R.; Spina, R.J.; King, D.S.; Rogers, M.A.; Brown, M.; Nemeth, P.M.; Holloszy, J.O. Skeletal muscle adaptations to endurance training in 60- to 70-yr-old men and women. J. Appl. Physiol. (1985) 1992, 72, 1780–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, D.N.; Sinning, W.E.; Walro, J.M.; Sieck, G.C.; Lemon, P.W. Oxidative capacity of human muscle fiber types: Effects of age and training status. J. Appl. Physiol. (1985) 1995, 78, 2033–2038. [Google Scholar] [CrossRef]
- Landers-Ramos, R.Q.; Prior, S.J. The Microvasculature and Skeletal Muscle Health in Aging. Exerc. Sport Sci. Rev. 2018, 46, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Gaster, M.; Poulsen, P.; Handberg, A.; Schroder, H.D.; Beck-Nielsen, H. Direct evidence of fiber type-dependent GLUT-4 expression in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E910–E916. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S.; Reggiani, C.; Mann, M. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef] [Green Version]
- Consitt, L.A.; Dudley, C.; Saxena, G. Impact of Endurance and Resistance Training on Skeletal Muscle Glucose Metabolism in Older Adults. Nutrients 2019, 11, 2636. [Google Scholar] [CrossRef] [Green Version]
- Tucker, M.Z.; Turcotte, L.P. Impaired fatty acid oxidation in muscle of aging rats perfused under basal conditions. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1102–E1109. [Google Scholar] [CrossRef] [Green Version]
- Tucker, M.Z.; Turcotte, L.P. Aging is associated with elevated muscle triglyceride content and increased insulin-stimulated fatty acid uptake. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E827–E835. [Google Scholar] [CrossRef] [Green Version]
- Volpi, E.; Mittendorfer, B.; Rasmussen, B.B.; Wolfe, R.R. The response of muscle protein anabolism to combined hyperaminoacidemia and glucose-induced hyperinsulinemia is impaired in the elderly. J. Clin. Endocrinol. Metab. 2000, 85, 4481–4490. [Google Scholar] [CrossRef] [Green Version]
- Paddon-Jones, D.; Sheffield-Moore, M.; Zhang, X.J.; Volpi, E.; Wolf, S.E.; Aarsland, A.; Ferrando, A.A.; Wolfe, R.R. Amino acid ingestion improves muscle protein synthesis in the young and elderly. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E321–E328. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Rasmussen, B.B. Skeletal muscle protein balance and metabolism in the elderly. Curr. Aging Sci. 2011, 4, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpi, E.; Sheffield-Moore, M.; Rasmussen, B.B.; Wolfe, R.R. Basal muscle amino acid kinetics and protein synthesis in healthy young and older men. JAMA 2001, 286, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Rennie, M.J. Anabolic resistance: The effects of aging, sexual dimorphism, and immobilization on human muscle protein turnover. Appl. Physiol. Nutr. Metab. 2009, 34, 377–381. [Google Scholar] [CrossRef]
- Cobley, J.N.; Sakellariou, G.K.; Murray, S.; Waldron, S.; Gregson, W.; Burniston, J.G.; Morton, J.P.; Iwanejko, L.A.; Close, G.L. Lifelong endurance training attenuates age-related genotoxic stress in human skeletal muscle. Longev. Healthspan 2013, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Cobley, J.N.; Sakellariou, G.K.; Owens, D.J.; Murray, S.; Waldron, S.; Gregson, W.; Fraser, W.D.; Burniston, J.G.; Iwanejko, L.A.; McArdle, A.; et al. Lifelong training preserves some redox-regulated adaptive responses after an acute exercise stimulus in aged human skeletal muscle. Free Radic. Biol. Med. 2014, 70, 23–32. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Deubel, S.; Jung, T.; Ost, M.; Klaus, S.; Grune, T.; Castro, J.P. Low proteasomal activity in fast skeletal muscle fibers is not associated with increased age-related oxidative damage. Exp. Gerontol. 2019, 117, 45–52. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Cogswell, A.M.; Stevens, R.J.; Hood, D.A. Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am. J. Physiol. 1993, 264, C383–C389. [Google Scholar] [CrossRef]
- Hood, D.A. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. (1985) 2001, 90, 1137–1157. [Google Scholar] [CrossRef]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. Appl. Physiol. (1985) 2013, 114, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Joseph, A.M.; Adhihetty, P.J.; Huang, J.H.; Saleem, A.; Uguccioni, G.; Hood, D.A. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany N. Y.) 2009, 1, 818–830. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.; Vitorino, R.; Alves, R.M.; Appell, H.J.; Powers, S.K.; Duarte, J.A.; Amado, F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 2010, 10, 3142–3154. [Google Scholar] [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Ma, C.; Li, Y.; Weisleder, N.; Rios, E.; Ma, J.; Zhou, J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 2011, 286, 32436–32443. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.R.; Cordova, A.; Valladares, D.; Llanos, P.; Hidalgo, C.; Gherardi, G.; De, S.D.; Mammucari, C.; Rizzuto, R.; Contreras-Ferrat, A.; et al. Mitochondrial Calcium Increase Induced by RyR1 and IP3R Channel Activation After Membrane Depolarization Regulates Skeletal Muscle Metabolism. Front. Physiol. 2018, 9, 791. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Dahl, R.; Larsen, S.; Dohlmann, T.L.; Qvortrup, K.; Helge, J.W.; Dela, F.; Prats, C. Three-dimensional reconstruction of the human skeletal muscle mitochondrial network as a tool to assess mitochondrial content and structural organization. Acta Physiol. (Oxf.) 2015, 213, 145–155. [Google Scholar] [CrossRef]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial Dynamics is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.; Ostojic, O.; Singh, K.; Joseph, A.M.; Hood, D.A. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 2013, 48, 963–970. [Google Scholar] [CrossRef]
- Nielsen, J.; Gejl, K.D.; Hey-Mogensen, M.; Holmberg, H.C.; Suetta, C.; Krustrup, P.; Elemans, C.P.H.; Ortenblad, N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 2017, 595, 2839–2847. [Google Scholar] [CrossRef]
- Hoppeler, H. Exercise-induced ultrastructural changes in skeletal muscle. Int. J. Sports Med. 1986, 7, 187–204. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Little, J.P.; Stokl, A.J.; Hettinga, B.P.; Akhtar, M.; Tarnopolsky, M.A. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J. Biol. Chem. 2011, 286, 10605–10617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Gurd, B.J. Deacetylation of PGC-1alpha by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [Green Version]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Marcinek, D.J.; Schenkman, K.A.; Ciesielski, W.A.; Lee, D.; Conley, K.E. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J. Physiol. 2005, 569, 467–473. [Google Scholar] [CrossRef]
- Masuyama, M.; Iida, R.; Takatsuka, H.; Yasuda, T.; Matsuki, T. Quantitative change in mitochondrial DNA content in various mouse tissues during aging. Biochim. Biophys. Acta 2005, 1723, 302–308. [Google Scholar] [CrossRef]
- Yeo, D.; Kang, C.; Gomez-Cabrera, M.C.; Vina, J.; Ji, L.L. Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo. Free Radic. Biol. Med. 2019, 130, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, M.; Brown, A.; Merrell, E.; DiSanto-Rose, M.; Rathmacher, J.A.; Reynolds, T.H. PKB signaling and atrogene expression in skeletal muscle of aged mice. J. Appl. Physiol. (1985) 2011, 111, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean, P.F.; Sgarioto, N.; Auger, M.J.; Vallee, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [Green Version]
- Koltai, E.; Hart, N.; Taylor, A.W.; Goto, S.; Ngo, J.K.; Davies, K.J.; Radak, Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R127–R134. [Google Scholar] [CrossRef] [Green Version]
- Pesce, V.; Cormio, A.; Fracasso, F.; Lezza, A.M.; Cantatore, P.; Gadaleta, M.N. Age-related changes of mitochondrial DNA content and mitochondrial genotypic and phenotypic alterations in rat hind-limb skeletal muscles. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 715–723. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Ritchie, D.; Thomas, M.M.; Wright, K.J.; Hepple, R.T. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell 2011, 10, 1047–1055. [Google Scholar] [CrossRef]
- Huang, J.H.; Joseph, A.M.; Ljubicic, V.; Iqbal, S.; Hood, D.A. Effect of age on the processing and import of matrix-destined mitochondrial proteins in skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.M.; Ljubicic, V.; Adhihetty, P.J.; Hood, D.A. Biogenesis of the mitochondrial Tom40 channel in skeletal muscle from aged animals and its adaptability to chronic contractile activity. Am. J. Physiol. Cell. Physiol. 2010, 298, C1308–C1314. [Google Scholar] [CrossRef]
- Bua, E.A.; McKiernan, S.H.; Wanagat, J.; McKenzie, D.; Aiken, J.M. Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J. Appl. Physiol. (1985) 2002, 92, 2617–2624. [Google Scholar] [CrossRef]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- McKiernan, S.H.; Colman, R.; Lopez, M.; Beasley, T.M.; Weindruch, R.; Aiken, J.M. Longitudinal analysis of early stage sarcopenia in aging rhesus monkeys. Exp. Gerontol. 2009, 44, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Boffoli, D.; Scacco, S.C.; Vergari, R.; Solarino, G.; Santacroce, G.; Papa, S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1994, 1226, 73–82. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Kent-Braun, J.A.; Ng, A.V. Skeletal muscle oxidative capacity in young and older women and men. J. Appl. Physiol. (1985) 2000, 89, 1072–1078. [Google Scholar] [CrossRef]
- Liu, D.; Sartor, M.A.; Nader, G.A.; Pistilli, E.E.; Tanton, L.; Lilly, C.; Gutmann, L.; IglayReger, H.B.; Visich, P.S.; Hoffman, E.P.; et al. Microarray analysis reveals novel features of the muscle aging process in men and women. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Lertwattanarak, R.; Lefort, N.; Molina-Carrion, M.; Joya-Galeana, J.; Bowen, B.P.; Garduno-Garcia, J.J.; Abdul-Ghani, M.; Richardson, A.; DeFronzo, R.A.; et al. Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 2011, 60, 2051–2060. [Google Scholar] [CrossRef] [Green Version]
- Lezza, A.M.; Pesce, V.; Cormio, A.; Fracasso, F.; Vecchiet, J.; Felzani, G.; Cantatore, P.; Gadaleta, M.N. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 in skeletal muscle from aged human subjects. FEBS Lett. 2001, 501, 74–78. [Google Scholar] [CrossRef]
- Melov, S.; Shoffner, J.M.; Kaufman, A.; Wallace, D.C. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 1995, 23, 4122–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, A.; Lee, C.C.; Vandiver, A.R.; Aiken, J.M.; McKenzie, D.; Hoang, A.; Allison, D.; Liu, N.; Wanagat, J. Mitochondrial DNA deletion mutations increase exponentially with age in human skeletal muscle. Aging Clin. Exp. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Hamadeh, M.J.; Kaczor, J.J.; Raha, S.; deBeer, J.; Tarnopolsky, M.A. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE 2010, 5, e10778. [Google Scholar] [CrossRef]
- Callahan, D.M.; Bedrin, N.G.; Subramanian, M.; Berking, J.; Ades, P.A.; Toth, M.J.; Miller, M.S. Age-related structural alterations in human skeletal muscle fibers and mitochondria are sex specific: Relationship to single-fiber function. J. Appl. Physiol. (1985) 2014, 116, 1582–1592. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [Green Version]
- Marzetti, E.; Leeuwenburgh, C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp. Gerontol. 2006, 41, 1234–1238. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Gijsen, A.P.; Saris, W.H.; Soeters, P.B.; Wagenmakers, A.J. Derangement in aerobic and anaerobic energy metabolism in skeletal muscle of critically ill and recovering rats. Biochim. Biophys. Acta 1996, 1315, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.L.; Robinson, M.M.; Nair, K.S. Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 2013, 24, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.N.; Chen, C.C.; Hood, D.A. Mitochondria, muscle health, and exercise with advancing age. Physiology (Bethesda) 2015, 30, 208–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beregi, E.; Regius, O.; Huttl, T.; Gobl, Z. Age-related changes in the skeletal muscle cells. Z. Gerontol. 1988, 21, 83–86. [Google Scholar] [PubMed]
- Aiken, J.; Bua, E.; Cao, Z.; Lopez, M.; Wanagat, J.; McKenzie, D.; McKiernan, S. Mitochondrial DNA deletion mutations and sarcopenia. Ann. N. Y. Acad. Sci. 2002, 959, 412–423. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, D.; Bua, E.; McKiernan, S.; Cao, Z.; Aiken, J.M. Mitochondrial DNA deletion mutations: A causal role in sarcopenia. Eur. J. Biochem. 2002, 269, 2010–2015. [Google Scholar] [CrossRef]
- Dodds, R.M.; Davies, K.; Granic, A.; Hollingsworth, K.G.; Warren, C.; Gorman, G.; Turnbull, D.M.; Sayer, A.A. Mitochondrial respiratory chain function and content are preserved in the skeletal muscle of active very old men and women. Exp. Gerontol. 2018, 113, 80–85. [Google Scholar] [CrossRef]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Grady, J.P.; Turnbull, D.M. Respiratory chain deficiency in aged spinal motor neurons. Neurobiol. Aging 2014, 35, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Pollock, N.; Staunton, C.A.; Vasilaki, A.; McArdle, A.; Jackson, M.J. Denervated muscle fibers induce mitochondrial peroxide generation in neighboring innervated fibers: Role in muscle aging. Free Radic. Biol. Med. 2017, 112, 84–92. [Google Scholar] [CrossRef]
- Joseph, A.M.; Adhihetty, P.J.; Wawrzyniak, N.R.; Wohlgemuth, S.E.; Picca, A.; Kujoth, G.C.; Prolla, T.A.; Leeuwenburgh, C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 2013, 8, e69327. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Calvani, R.; Lorenzi, M.; Menghi, A.; Galli, M.; Vitiello, R.; Randisi, F.; Bernabei, R.; Landi, F.; Marzetti, E. Mitochondrial dynamics signaling is shifted toward fusion in muscles of very old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp. Gerontol. 2017, 96, 63–67. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Ichim, G.; Tait, S.W.; Passos, J.F. Depletion of mitochondria in mammalian cells through enforced mitophagy. Nat. Protoc. 2017, 12, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea, D.M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derbre, F.; Gomez-Cabrera, M.C.; Nascimento, A.L.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Tresguerres, J.A.; Fuentes, T.; Gratas-Delamarche, A.; Monsalve, M.; Vina, J. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1alpha to exercise training. Age (Dordr.) 2012, 34, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, M.; Miyashita, K.; Hagiwara, A.; Wakino, S.; Inoue, H.; Fujii, K.; Fujii, C.; Endo, S.; Uto, A.; Mitsuishi, M.; et al. Ghrelin treatment improves physical decline in sarcopenia model mice through muscular enhancement and mitochondrial activation. Endocr. J. 2017, 64, S47–S51. [Google Scholar] [CrossRef] [Green Version]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musaro, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ’exercise mimetic’ in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef]
- Vinel, C.; Lukjanenko, L.; Batut, A.; Deleruyelle, S.; Pradere, J.P.; Le, G.S.; Dortignac, A.; Geoffre, N.; Pereira, O.; Karaz, S.; et al. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018, 24, 1360–1371. [Google Scholar] [CrossRef]
- Kim, C.; Hwang, J.K. The 5,7-Dimethoxyflavone Suppresses Sarcopenia by Regulating Protein Turnover and Mitochondria Biogenesis-Related Pathways. Nutrients 2020, 12, 1079. [Google Scholar] [CrossRef]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; Lastayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, C422–C430. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picca, A.; Beli, R.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Bucci, C.; Guerra, F.; Marzetti, E. Older Adults with Physical Frailty and Sarcopenia Show Increased Levels of Circulating Small Extracellular Vesicles with a Specific Mitochondrial Signature. Cells 2020, 9, 973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Primeau, A.J.; Adhihetty, P.J.; Hood, D.A. Apoptosis in heart and skeletal muscle. Can. J. Appl. Physiol. 2002, 27, 349–395. [Google Scholar] [CrossRef]
- Mayer, B.; Oberbauer, R. Mitochondrial regulation of apoptosis. News Physiol. Sci. 2003, 18, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef] [Green Version]
- Cande, C.; Vahsen, N.; Garrido, C.; Kroemer, G. Apoptosis-inducing factor (AIF): Caspase-independent after all. Cell Death Differ. 2004, 11, 591–595. [Google Scholar] [CrossRef]
- Dirks, A.; Leeuwenburgh, C. Apoptosis in skeletal muscle with aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R519–R527. [Google Scholar] [CrossRef] [Green Version]
- Phillips, T.; Leeuwenburgh, C. Muscle fiber specific apoptosis and TNF-alpha signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 2005, 19, 668–670. [Google Scholar] [CrossRef]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef]
- Song, W.; Kwak, H.B.; Lawler, J.M. Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxid. Redox Signal. 2006, 8, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Adhihetty, P.J.; Ljubicic, V.; Hood, D.A. Effect of chronic contractile activity on SS and IMF mitochondrial apoptotic susceptibility in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E748–E755. [Google Scholar] [CrossRef]
- Salucci, S.; Battistelli, M.; Baldassarri, V.; Burini, D.; Falcieri, E.; Burattini, S. Melatonin prevents mitochondrial dysfunctions and death in differentiated skeletal muscle cells. Microsc. Res. Tech. 2017, 80, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Sayed, R.K.A.; Fernandez-Ortiz, M.; Diaz-Casado, M.E.; Rusanova, I.; Rahim, I.; Escames, G.; Lopez, L.C.; Mokhtar, D.M.; Acuna-Castroviejo, D. The Protective Effect of Melatonin against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1330–1338. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Species | Age Groups Studied | Relevant Finding in Old Skeletal Muscle | Reference |
|---|---|---|---|
| C57Bl/6 Mice | 7 vs. 30 months old | ↓ mitochondrial coupling | [80] |
| C57Bl/6 Mice | 2–15 months old | ↑ TFAM and mtDNA content | [81] |
| C57Bl/6 Mice | 2 vs. 24 months old | ↑ mitophagy ↑ mitochondrial fission | [82] |
| C57Bl/6 Mice | 11–13 vs. 25–27 months old | ↓ autophagy | [83] |
| C5l/6 Mice | 3 vs. 18 months old | ↓ mitochondrial content ↓ mitochondrial biogenesis ↑ mitophagy flux | [84] |
| Mice | 8–12 vs. 88–96 weeks old | ↑ mitochondrial fusion index | [85] |
| Wistar Rats | 3 vs. 26 months old | ↓ mitochondrial mass ↓ PGC-1α protein ↑ Fis1 and Mfs1 proteins | [86] |
| Wistar Rats | 3–28 months old | ↑ mtDNA deletions ↓ respiratory enzymes | [87] |
| Fischer 344 Brown Norway Rats | 5 vs. 35 months old | ↓ mitochondrial size ↑ mitochondrial fission proteins | [67] |
| Fischer 344 Brown Norway Rats | 6 vs. 24 months old | ↓ autophagy | [88] |
| Fischer 344 Brown Norway Rats | 6 vs. 36 months old | ↓ mitochondrial content ↑ mitochondrial ROS production ↑ cytochrome c and endonuclease G release | [56] |
| Fischer 344 Brown Norway Rats | 8–10 vs. 35–36 months old | ↓ OXPHOS proteins, CS activity, state III respiration, mPTP function ↑ free radical leak | [89] |
| Fischer 344 Brown Norway Rats | 6 vs. 35–38 months old | =mitochondrial protein import machinery | [90] |
| Fischer 344 Brown Norway Rats | 6 vs. 36 months old | ↑ protein assembly =protein import | [91] |
| Fischer 344 Brown Norway Rats | 5, 18, 36 months old | ↑ ETC abnormalities | [92] |
| Fischer 344 Brown Norway Rats | 5–6 vs. 35–36 months old | ↑ mitophagy flux | [93] |
| Rhesus Monkeys | 6, 9, 12 years old | ↑ enzyme abnormalities ↑ mtDNA deletion mutations | [94] |
| Humans | 17–91 years old | ↓ respiratory activity of complex I, II, and IV | [95] |
| Humans | 18–89 years old | ↓ mtDNA, mRNA, and mitochondrial proteins ↓ mitochondrial ATP production | [96] |
| Humans | 29–80 years old | ↓ oxidative capacity | [97] |
| Humans | 22–75 years old | ↓ mitochondrial transcriptome | [98] |
| Humans | 25–72 years old | ↓ ETC proteins ↓ ROS production | [99] |
| Humans | 21–88 years old | ↑ TFAM mRNA and protein | [100] |
| Humans | 20–71 years old | ↑ mtDNA rearrangements | [101] |
| Humans | 49–93 years old | ↑ mtDNA deletion mutations | [102] |
| Humans | 20–80 years old | ↑ mtDNA deletions | [103] |
| Humans | 20–75 years old | ↓ mitochondrial enzymes activity ↓ mitochondrial biogenesis | [104] |
| Humans | 21–75 years old | ↓ IMF mitochondrial size | [105] |
| Humans | 22–82 years old | ↓ mitochondrial respiration ↓ PGC-1α, COX I, and OPA proteins ↑ mitochondrial protein import machinery | [106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellanti, F.; Lo Buglio, A.; Vendemiale, G. Mitochondrial Impairment in Sarcopenia. Biology 2021, 10, 31. https://doi.org/10.3390/biology10010031
Bellanti F, Lo Buglio A, Vendemiale G. Mitochondrial Impairment in Sarcopenia. Biology. 2021; 10(1):31. https://doi.org/10.3390/biology10010031
Chicago/Turabian StyleBellanti, Francesco, Aurelio Lo Buglio, and Gianluigi Vendemiale. 2021. "Mitochondrial Impairment in Sarcopenia" Biology 10, no. 1: 31. https://doi.org/10.3390/biology10010031