A Destruction Model of the Vascular and Lymphatic Systems in the Emergence of Psychiatric Symptoms



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Main Text
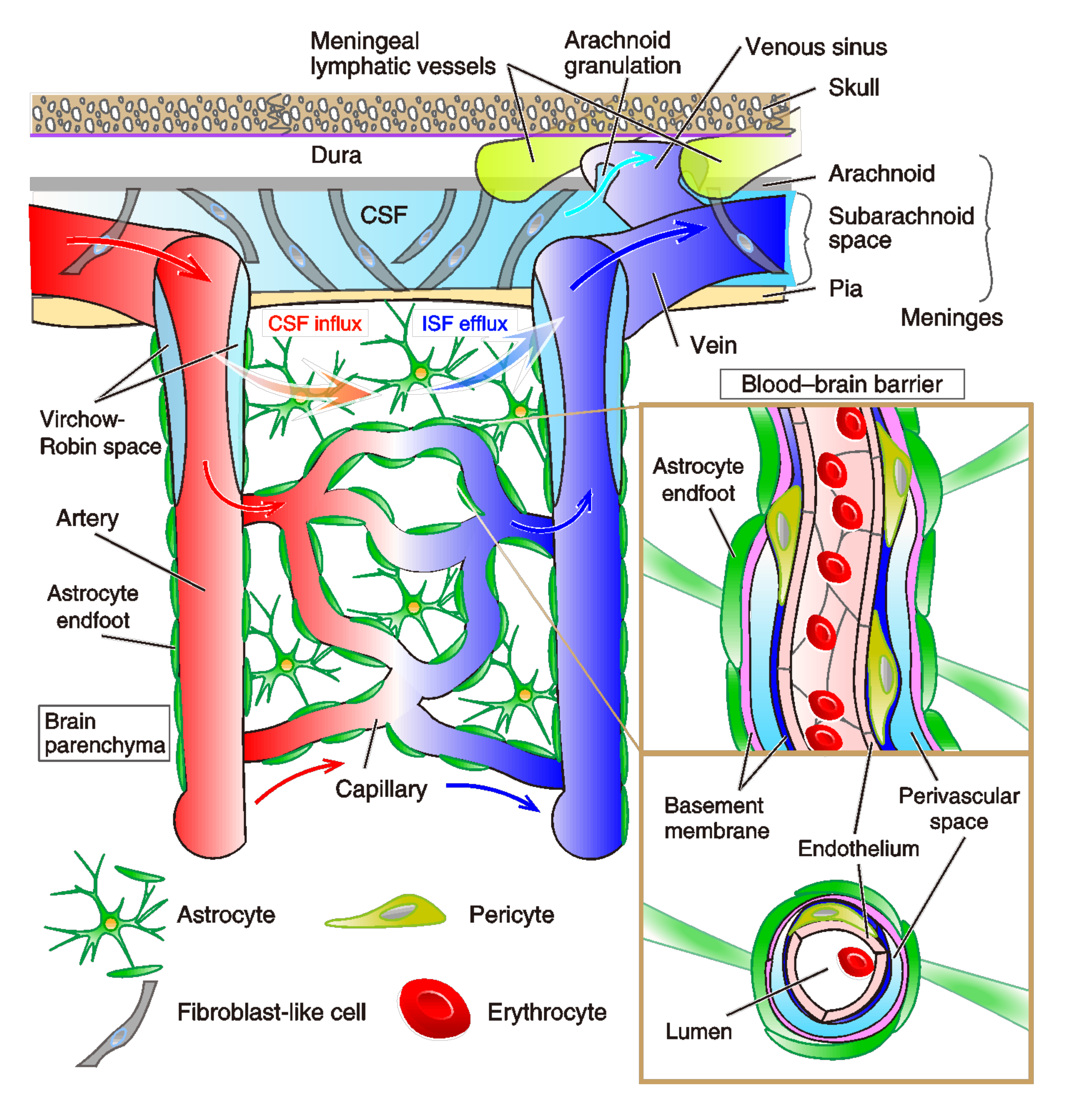
2.1. Vascular Lymphatic System in the Brain
2.1.1. Brain Vasculature System
2.1.2. Characteristics of Meningeal Lymphatic System
2.1.3. Lymphatic Vasculogenesis
2.2. Glia-Lymphatic System (Glymphatic System)
2.2.1. Glymphatic System
2.2.2. Astrocytic Contribution and AQP-4 Function
2.2.3. Potential Pathophysiology of the Glymphatic System
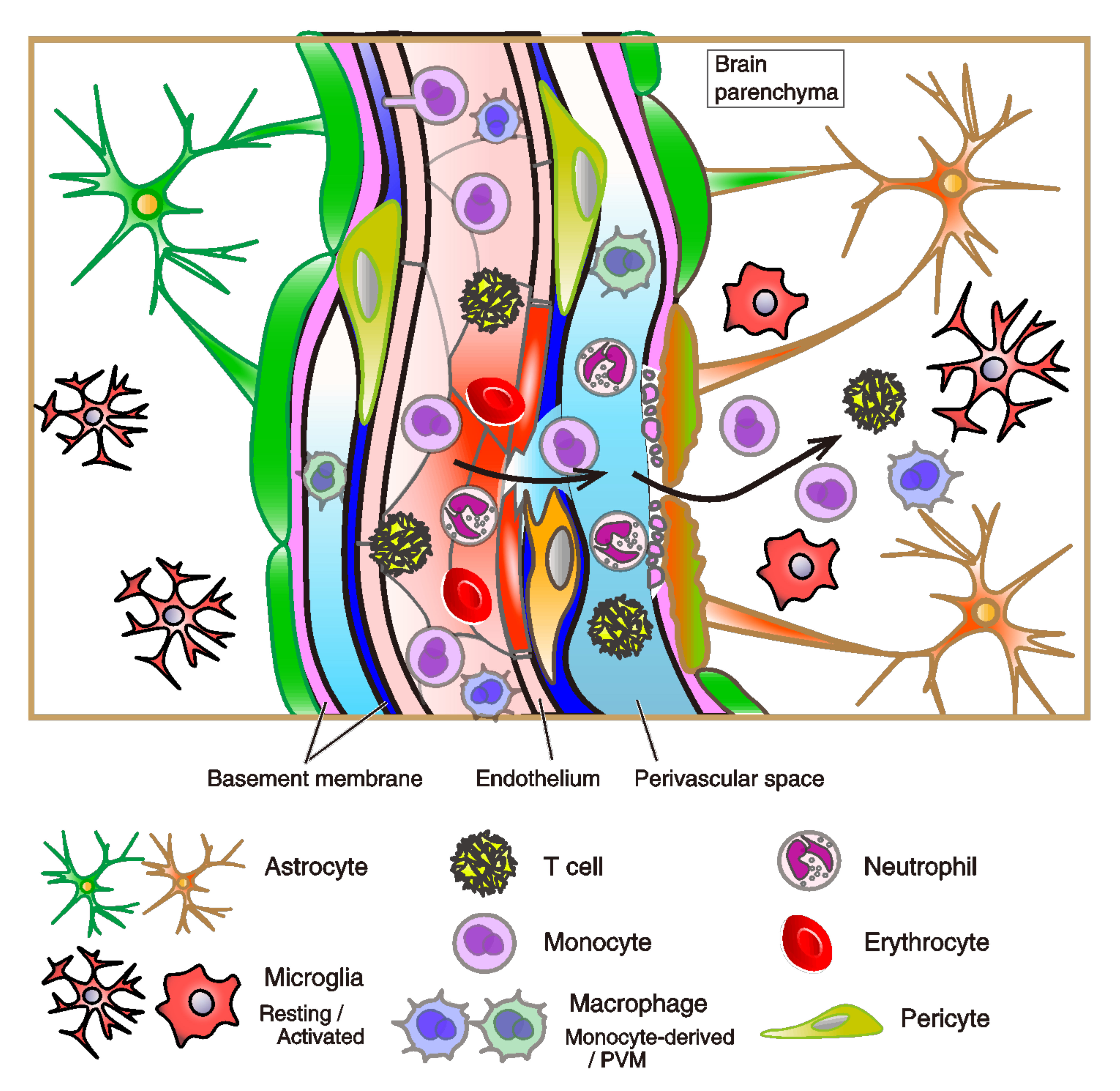
2.3. Infiltration of Immune Cells to Brain across Vasculature System
2.3.1. Immune Privilege
2.3.2. Glial Cell-Characteristics of the Brain and the Regulation by Immunity
2.3.3. Infiltration of Immune Cells to Brain Parenchyma
2.3.4. The Structure and Dysfunction of the BBB
2.4. Implications of the Inflammation and Immunity for the Pathophysiology of Psychiatric Disorders
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tzour, A.; Leibovich, H.; Barkai, O.; Biala, Y.; Lev, S.; Yaari, Y.; Binshtok, A.M. KV 7/M channels as targets for lipopolysaccharide-induced inflammatory neuronal hyperexcitability. J. Physiol. 2017, 595, 713–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alitalo, K. The lymphatic vasculature in disease. Nat. Med. 2011, 17, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147.ra111. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Mascagni, P. Vasorum Lymphaticorum Corporis Humani Historia et Ichnographia; Ex Typogr. Pazzini Carli: Siena, Italy, 1787; pp. 1522–1867. [Google Scholar]
- Sandrone, S.; Moreno-Zambrano, D.; Kipnis, J.; van Gijn, J. A (delayed) history of the brain lymphatic system. Nat. Med. 2019, 25, 538–540. [Google Scholar] [CrossRef]
- Absinta, M.; Ha, S.K.; Nair, G.; Sati, P.; Luciano, N.J.; Palisoc, M.; Louveau, A.; Zaghloul, K.A.; Pittaluga, S.; Kipnis, J.; et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife 2017, 6, e29738. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Ohtsuki, G.; Shishikura, M.; Ozaki, A. Synergistic excitability plasticity in cerebellar functioning. FEBS J. 2020, 287, 4557–4593. [Google Scholar] [CrossRef]
- Dietz, A.G.; Goldman, S.A.; Nedergaard, M. Glial cells in schizophrenia: A unified hypothesis. Lancet Psychiatry 2020, 7, 272–281. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Da Mesquita, S.; Kipnis, J. Lymphatics in Neurological Disorders: A Neuro-Lympho-Vascular Component of Multiple Sclerosis and Alzheimer’s Disease? Neuron 2016, 91, 957–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Ahn, J.H.; Cho, H.; Kim, J.H.; Kim, S.H.; Ham, J.S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.H.; Hong, Y.K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef]
- Miyawaki, T.; Morikawa, S.; Susaki, E.A.; Nakashima, A.; Takeuchi, H.; Yamaguchi, S.; Ueda, H.R.; Ikegaya, Y. Visualization and molecular characterization of whole-brain vascular networks with capillary resolution. Nat. Commun. 2020, 11, 1104. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Kerjaschki, D. The lymphatic vasculature revisited. J. Clin. Investig. 2014, 124, 874–877. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Oliver, G. Current views on the function of the lymphatic vasculature in health and disease. Genes. Dev. 2010, 24, 2115–2126. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Kiviniemi, V.; Wang, X.; Korhonen, V.; Keinänen, T.; Tuovinen, T.; Autio, J.; LeVan, P.; Keilholz, S.; Zang, Y.-F.; Hennig, J.; et al. Ultra-fast magnetic resonance encephalography of physiological brain activity—Glymphatic pulsation mechanisms? J. Cereb. Blood Flow Metab. 2016, 36, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Wichterle, H.; Gonzalez-Perez, O.; Cholfin, J.A.; Yamada, M.; Spassky, N.; Murcia, N.S.; Garcia-Verdugo, J.M.; Marin, O.; Rubenstein, J.L.R.; et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science 2006, 311, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Nagira, M.; Kitaura, M.; Imagawa, N.; Imai, T.; Yoshie, O. Secondary lymphoid-tissue chemokine is a functional ligand for the CC chemokine receptor CCR7. J. Biol. Chem. 1998, 273, 7118–7122. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Palermo, B.; Lenig, D.; Miettinen, M.; Matikainen, S.; Julkunen, I.; Forster, R.; Burgstahler, R.; Lipp, M.; Lanzavecchia, A. Distinct patterns and kinetics of chemokine production regulate dendritic cell function. Eur. J. Immunol. 1999, 29, 1617–1625. [Google Scholar] [CrossRef]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Schulte-Merker, S.; Sabine, A.; Petrova, T.V. Lymphatic vascular morphogenesis in development, physiology, and disease. J. Cell Biol. 2011, 193, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, L.; Xu, H.; Xing, L.; Zhuang, Z.; Zheng, Y.; Li, X.; Wang, C.; Chen, S.; Guo, Z.; et al. Meningeal lymphatics clear erythrocytes that arise from subarachnoid hemorrhage. Nat. Commun. 2020, 11, 3159. [Google Scholar] [CrossRef]
- Schmahmann, J.D. Disorders of the cerebellum: Ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 367–378. [Google Scholar] [CrossRef]
- Tsai, P.T.; Hull, C.; Chu, Y.X.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, A.; Yamawaki, Y.; Ohtsuki, G. Psychosis symptoms following aberrant immunity in the brain. Neural. Regen. Res. 2021, 16, 512–513. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, G.; Kawaguchi, S.Y.; Mishina, M.; Hirano, T. Enhanced inhibitory synaptic transmission in the cerebellar molecular layer of the GluRδ2 knock-out mouse. J. Neurosci. 2004, 24, 10900–10907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, E.K.; Sekerková, G.; Ohtsuki, G.; Aldinger, K.A.; Chizhikov, V.V.; Hansel, C.; Mugnaini, E.; Millen, K.J. The Spontaneous Ataxic Mouse Mutant Tippy is Characterized by a Novel Purkinje Cell Morphogenesis and Degeneration Phenotype. Cerebellum 2015, 14, 292–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.F.; Beevis, J.C.; Empson, R.M. Essential Tremor—A cerebellar driven disorder? Neuroscience 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K. Morphology of cerebral circulation. Kawasaki Igakkai Shi Lib. Arts Sci. 2011, 37, 19–29. [Google Scholar]
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.S.; Dillard, M.E.; Lagutin, O.V.; Lin, F.; Tsai, F.; Tsai, M.J.; Samokhvalov, I.M.; Oliver, G. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes. Dev. 2007, 21, 2422–2432. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Corral, I.; Ulvmar, M.H.; Stanczuk, L.; Tatin, F.; Kizhatil, K.; John, S.W.; Alitalo, K.; Ortega, S.; Makinen, T. Nonvenous origin of dermal lymphatic vasculature. Circ. Res. 2015, 116, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- François, M.; Caprini, A.; Hosking, B.; Orsenigo, F.; Wilhelm, D.; Browne, C.; Paavonen, K.; Karnezis, T.; Shayan, R.; Downes, M.; et al. Sox18 induces development of the lymphatic vasculature in mice. Nature 2008, 456, 643–647. [Google Scholar] [CrossRef]
- Petrova, T.V.; Mäkinen, T.; Mäkelä, T.P.; Saarela, J.; Virtanen, I.; Ferrell, R.E.; Finegold, D.N.; Kerjaschki, D.; Ylä-Herttuala, S.; Alitalo, K. Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J. 2002, 21, 4593–4599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.J.; Chen, X.; Qin, J.; Hong, Y.K.; Tsai, M.J.; Tsai, S.Y. Direct transcriptional regulation of neuropilin-2 by COUP-TFII modulates multiple steps in murine lymphatic vessel development. J. Clin. Investig. 2010, 120, 1694–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoppmann, S.F.; Birner, P.; Stöckl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Kerjaschki, D.; Huttary, N.; Raab, I.; Regele, H.; Bojarski-Nagy, K.; Bartel, G.; Kröber, S.M.; Greinix, H.; Rosenmaier, A.; Karlhofer, F.; et al. Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nat. Med. 2006, 12, 230–234. [Google Scholar] [CrossRef]
- Maruyama, K.; Ii, M.; Cursiefen, C.; Jackson, D.G.; Keino, H.; Tomita, M.; Van Rooijen, N.; Takenaka, H.; D’Amore, P.A.; Stein-Streilein, J.; et al. Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J. Clin. Investig. 2005, 115, 2363–2372. [Google Scholar] [CrossRef]
- Angeli, V.; Ginhoux, F.; Llodrà, J.; Quemeneur, L.; Frenette, P.S.; Skobe, M.; Jessberger, R.; Merad, M.; Randolph, G.J. B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity 2006, 24, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Kataru, R.P.; Kim, H.; Jang, C.; Choi, D.K.; Koh, B.I.; Kim, M.; Gollamudi, S.; Kim, Y.K.; Lee, S.H.; Koh, G.Y. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity 2011, 34, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.; Rayasam, A.; Kijak, J.A.; Choi, Y.H.; Harding, J.S.; Marcus, S.A.; Karpus, W.J.; Sandor, M.; Fabry, Z. Neuroinflammation-induced lymphangiogenesis near the cribriform plate contributes to drainage of CNS-derived antigens and immune cells. Nat. Commun. 2019, 10, 229. [Google Scholar] [CrossRef]
- Sloan, S.A.; Barres, B.A. Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr. Opin. Neurobiol. 2014, 27, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Ho, M.S.; Vardjan, N.; Zorec, R.; Parpura, V. General Pathophysiology of Astroglia. Adv. Exp. Med. Biol. 2019, 1175, 149–179. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006, 9, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008, 19, 1–5. [Google Scholar] [CrossRef]
- Desai, B.; Hsu, Y.; Schneller, B.; Hobbs, J.G.; Mehta, A.I.; Linninger, A. Hydrocephalus: The role of cerebral aquaporin-4 channels and computational modeling considerations of cerebrospinal fluid. Neurosurg. Focus. 2016, 41, E8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestre, H.; Mori, Y.; Nedergaard, M. The Brain’s Glymphatic System: Current Controversies. Trends Neurosci. 2020, 43, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Barker, A.J.; Ullian, E.M. New roles for astrocytes in developing synaptic circuits. Commun. Integr. Biol. 2008, 1, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Ikeshima-Kataoka, H. Neuroimmunological Implications of AQP4 in Astrocytes. Int. J. Mol. Sci. 2016, 17, 1306. [Google Scholar] [CrossRef]
- Ishibashi, T.; Dakin, K.A.; Stevens, B.; Lee, P.R.; Kozlov, S.V.; Stewart, C.L.; Fields, R.D. Astrocytes promote myelination in response to electrical impulses. Neuron 2006, 49, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Pekny, M.; Pekna, M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta 2016, 1862, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [Green Version]
- Harrison, I.F.; Siow, B.; Akilo, A.B.; Evans, P.G.; Ismail, O.; Ohene, Y.; Nahavandi, P.; Thomas, D.L.; Lythgoe, M.F.; Wells, J.A. Non-invasive imaging of CSF-mediated brain clearance pathways via assessment of perivascular fluid movement with diffusion tensor MRI. eLife 2018, 7, e34028. [Google Scholar] [CrossRef]
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef]
- Nedergaard, M. Neuroscience. Garbage truck of the brain. Science 2013, 340, 1529–1530. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Hablitz, L.M.; Vinitsky, H.S.; Sun, Q.; Stæger, F.F.; Sigurdsson, B.; Mortensen, K.N.; Lilius, T.O.; Nedergaard, M. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci. Adv. 2019, 5, eaav5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, J.; Davoodi-Bojd, E.; Fahmy, L.M.; Zhang, L.; Ding, G.; Hu, J.; Zhang, Z.; Chopp, M.; Jiang, Q. Magnetic Resonance Imaging and Modeling of the Glymphatic System. Diagnostics 2020, 10, 344. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, J.A.; Szu, J.I.; Binder, D.K. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res. Bull. 2018, 136, 118–129. [Google Scholar] [CrossRef]
- Silverberg, G.D.; Heit, G.; Huhn, S.; Jaffe, R.A.; Chang, S.D.; Bronte-Stewart, H.; Rubenstein, E.; Possin, K.; Saul, T.A. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology 2001, 57, 1763–1766. [Google Scholar] [CrossRef]
- Preston, J.E. Ageing choroid plexus-cerebrospinal fluid system. Microsc. Res. Tech. 2001, 52, 31–37. [Google Scholar] [CrossRef]
- Tuovinen, T.; Kananen, J.; Rajna, Z.; Lieslehto, J.; Korhonen, V.; Rytty, R.; Mattila, H.; Huotari, N.; Raitamaa, L.; Helakari, H.; et al. The variability of functional MRI brain signal increases in Alzheimer’s disease at cardiorespiratory frequencies. Sci. Rep. 2020, 10, 21559. [Google Scholar] [CrossRef]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, Ø.; Laake, P.; Klungland, A.; Thorén, A.E.; Burkhardt, J.M.; Ottersen, O.P.; et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [Green Version]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Zeppenfeld, D.M.; Venkataraman, A.; Plog, B.A.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 2013, 33, 18190–18199. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Suh, M.; Sarker, A.; Choi, Y. Brain Glymphatic/Lymphatic Imaging by MRI and PET. Nucl. Med. Mol. Imaging 2020, 54, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Gaberel, T.; Gakuba, C.; Goulay, R.; Martinez De Lizarrondo, S.; Hanouz, J.L.; Emery, E.; Touze, E.; Vivien, D.; Gauberti, M. Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: A new target for fibrinolysis? Stroke 2014, 45, 3092–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Taoka, T.; Naganawa, S. Gadolinium-based Contrast Media, Cerebrospinal Fluid and the Glymphatic System: Possible Mechanisms for the Deposition of Gadolinium in the Brain. Magn. Reson. Med. Sci. 2018, 17, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Ohki, K.; Chung, S.; Ch’ng, Y.H.; Kara, P.; Reid, R.C. Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex. Nature 2005, 433, 597–603. [Google Scholar] [CrossRef]
- Ohtsuki, G.; Nishiyama, M.; Yoshida, T.; Murakami, T.; Histed, M.; Lois, C.; Ohki, K. Similarity of visual selectivity among clonally related neurons in visual cortex. Neuron 2012, 75, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.È.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef]
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Abrahao, A.; Heyn, C.C.; Bethune, A.J.; Huang, Y.; Pople, C.B.; Aubert, I.; Hamani, C.; Zinman, L.; Hynynen, K.; et al. Glymphatics Visualization after Focused Ultrasound-Induced Blood-Brain Barrier Opening in Humans. Ann. Neurol. 2019, 86, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Messing, A.; Brenner, M.; Feany, M.B.; Nedergaard, M.; Goldman, J.E. Alexander disease. J. Neurosci. 2012, 32, 5017–5023. [Google Scholar] [CrossRef] [PubMed]
- Messing, A. Refining the concept of GFAP toxicity in Alexander disease. J. Neurodev. Disord. 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, R.A.; Brenner, M.; Goldman, J.E.; Messing, A. GFAP and its role in Alexander disease. Exp. Cell Res. 2007, 313, 2077–2087. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, T.L.; Gaeta, S.A.; Smith, M.A.; Johnson, D.A.; Johnson, J.A.; Messing, A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum. Mol. Genet. 2005, 14, 2443–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, V.C.; Tian, R.; Long, H.; Der Perng, M.; Brenner, M.; Quinlan, R.A.; Goldman, J.E. Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J. Cell Sci. 2005, 118, 2057–2065. [Google Scholar] [CrossRef] [Green Version]
- Eid, T.; Lee, T.-S.W.; Thomas, M.J.; Amiry-Moghaddam, M.; Bjørnsen, L.P.; Spencer, D.D.; Agre, P.; Ottersen, O.P.; de Lanerolle, N.C. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Oberheim, N.A.; Tian, G.-F.; Han, X.; Peng, W.; Takano, T.; Ransom, B.; Nedergaard, M. Loss of astrocytic domain organization in the epileptic brain. J. Neurosci. 2008, 28, 3264–3276. [Google Scholar] [CrossRef] [Green Version]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Szternfeld, R.D.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Kananen, J.; Helakari, H.; Korhonen, V.; Huotari, N.; Järvelä, M.; Raitamaa, L.; Raatikainen, V.; Rajna, Z.; Tuovinen, T.; Nedergaard, M.; et al. Respiratory-related brain pulsations are increased in epilepsy-a two-centre functional MRI study. Brain Commun. 2020, 2, fcaa076. [Google Scholar] [CrossRef]
- Niederkorn, J.Y. See no evil, hear no evil, do no evil: The lessons of immune privilege. Nat. Immunol. 2006, 7, 354–359. [Google Scholar] [CrossRef]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef] [PubMed]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [Green Version]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. Control of autoimmune CNS inflammation by astrocytes. Semin. Immunopathol. 2015, 37, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Nave, K.A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Lee, P.R.; Fields, R.D. Control of local protein synthesis and initial events in myelination by action potentials. Science 2011, 333, 1647–1651. [Google Scholar] [CrossRef] [Green Version]
- Makinodan, M.; Rosen, K.M.; Ito, S.; Corfas, G. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 2012, 337, 1357–1360. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, Y.; Wakabayashi, T.; Mori, T.; Koike, T.; Yao, I.; Tsuda, M.; Honke, K.; Gotoh, H.; Ono, K.; Yamada, H. Sulfatide species with various fatty acid chains in oligodendrocytes at different developmental stages determined by imaging mass spectrometry. J. Neurochem. 2017, 140, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossad, O.; Erny, D. The microbiota-microglia axis in central nervous system disorders. Brain Pathol. 2020, 30, 1159–1177. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef]
- Greter, M.; Merad, M. Regulation of microglia development and homeostasis. Glia 2013, 61, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kim, M.; Imai, H.; Itakura, Y.; Ohtsuki, G. Microglia-Triggered Plasticity of Intrinsic Excitability Modulates Psychomotor Behaviors in Acute Cerebellar Inflammation. Cell Rep. 2019, 28, 2923–2938. [Google Scholar] [CrossRef]
- Zhang, J.; Malik, A.; Choi, H.B.; Ko, R.W.Y.; Dissing-Olesen, L.; MacVicar, B.A. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron 2014, 82, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernández-Klett, F.; Lin, G.; Sagar, D.M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; Staszewski, O.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas, A.S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada, G.F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Stoessel, M.B.; Majewska, A.K. Cerebellar microglia are more immunologically vigilant. Trends Neurosci. in revision.
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar, S.E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; Heiland, D.H.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar, S.C.; Nessler, S.; Kunz, P.; van Loo, G.; Coenen, V.A.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell 2020, 27, 951–961. [Google Scholar] [CrossRef]
- Lapenna, A.; De Palma, M.; Lewis, C.E. Perivascular macrophages in health and disease. Nat. Rev. Immunol. 2018, 18, 689–702. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Merlini, M.; Späni, C.; Gericke, C.; Schweizer, N.; Enzmann, G.; Engelhardt, B.; Kulic, L.; Suter, T.; Nitsch, R.M. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav. Immun. 2016, 54, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Ten Berge, I.J.M.; van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Debes, G.F.; Arnold, C.N.; Young, A.J.; Krautwald, S.; Lipp, M.; Hay, J.B.; Butcher, E.C. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 2005, 6, 889–894. [Google Scholar] [CrossRef] [Green Version]
- Bromley, S.K.; Thomas, S.Y.; Luster, A.D. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 2005, 6, 895–901. [Google Scholar] [CrossRef]
- Ohl, L.; Mohaupt, M.; Czeloth, N.; Hintzen, G.; Kiafard, Z.; Zwirner, J.; Blankenstein, T.; Henning, G.; Förster, R. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 2004, 21, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Jackson, D.G. The chemokine CX3CL1 promotes trafficking of dendritic cells through inflamed lymphatics. J. Cell Sci. 2013, 126, 5259–5270. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.M.; Liu, Y.; Qian, Z.M.; Luo, Q.Q.; Ke, Y. CX3CL1/CX3CR1 Axis Plays a Key Role in Ischemia-Induced Oligodendrocyte Injury via p38MAPK Signaling Pathway. Mol. Neurobiol. 2016, 53, 4010–4018. [Google Scholar] [CrossRef]
- Brown, M.; Johnson, L.A.; Leone, D.A.; Majek, P.; Vaahtomeri, K.; Senfter, D.; Bukosza, N.; Schachner, H.; Asfour, G.; Langer, B.; et al. Lymphatic exosomes promote dendritic cell migration along guidance cues. J. Cell Biol. 2018, 217, 2205–2221. [Google Scholar] [CrossRef] [PubMed]
- Sawa, Y.; Tsuruga, E. The expression of E-selectin and chemokines in the cultured human lymphatic endothelium with lipopolysaccharides. J. Anat. 2008, 212, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Vigl, B.; Aebischer, D.; Nitschké, M.; Iolyeva, M.; Röthlin, T.; Antsiferova, O.; Halin, C. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 2011, 118, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Banerji, S.; Lawrance, W.; Gileadi, U.; Prota, G.; Holder, K.A.; Roshorm, Y.M.; Hanke, T.; Cerundolo, V.; Gale, N.W.; et al. Dendritic cells enter lymph vessels by hyaluronan-mediated docking to the endothelial receptor LYVE-1. Nat. Immunol. 2017, 18, 762–770. [Google Scholar] [CrossRef]
- Mondor, I.; Baratin, M.; Lagueyrie, M.; Saro, L.; Henri, S.; Gentek, R.; Suerinck, D.; Kastenmuller, W.; Jiang, J.X.; Bajénoff, M. Lymphatic Endothelial Cells Are Essential Components of the Subcapsular Sinus Macrophage Niche. Immunity 2019, 50, 1453–1466.e4. [Google Scholar] [CrossRef]
- Kiviniemi, V.; Korhonen, V.; Kortelainen, J.; Rytky, S.; Keinänen, T.; Tuovinen, T.; Isokangas, M.; Sonkajärvi, E.; Siniluoto, T.; Nikkinen, J.; et al. Real-time monitoring of human blood-brain barrier disruption. PLoS ONE 2017, 12, e0174072. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Fong, C.W. Permeability of the Blood-Brain Barrier: Molecular Mechanism of Transport of Drugs and Physiologically Important Compounds. J. Membr. Biol. 2015, 248, 651–669. [Google Scholar] [CrossRef]
- Caprifico, A.E.; Foot, P.J.S.; Polycarpou, E.; Calabrese, G. Overcoming the Blood-Brain Barrier: Functionalised Chitosan Nanocarriers. Pharmaceutics 2020, 12, 1013. [Google Scholar] [CrossRef]
- Aryal, M.; Zhou, Q.; Rosenthal, E.L.; Airan, R.D. Noninvasive Ultrasonic Glymphatic Induction Enhances Intrathecal Drug Delivery. bioRxiv 2020. [Google Scholar] [CrossRef]
- Haumann, R.; Videira, J.C.; Kaspers, G.J.L.; van Vuurden, D.G.; Hulleman, E. Overview of Current Drug Delivery Methods Across the Blood-Brain Barrier for the Treatment of Primary Brain Tumors. CNS Drugs 2020, 34, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell. 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [Green Version]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef] [Green Version]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Cerebrovascular effects of apolipoprotein E: Implications for Alzheimer disease. JAMA Neurol. 2013, 70, 440–444. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef]
- Santello, M.; Bezzi, P.; Volterra, A. TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011, 69, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Bellinger, F.P.; Madamba, S.; Siggins, G.R. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993, 628, 227–234. [Google Scholar] [CrossRef]
- Schneider, H.; Pitossi, F.; Balschun, D.; Wagner, A.; del Rey, A.; Besedovsky, H.O. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc. Natl. Acad. Sci. USA 1998, 95, 7778–7783. [Google Scholar] [CrossRef] [Green Version]
- Favrais, G.; van de Looij, Y.; Fleiss, B.; Ramanantsoa, N.; Bonnin, P.; Stoltenburg-Didinger, G.; Lacaud, A.; Saliba, E.; Dammann, O.; Gallego, J.; et al. Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 2011, 70, 550–565. [Google Scholar] [CrossRef]
- Pang, Y.; Campbell, L.; Zheng, B.; Fan, L.; Cai, Z.; Rhodes, P. Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neuroscience 2010, 166, 464–475. [Google Scholar] [CrossRef]
- Gibson, E.M.; Nagaraja, S.; Ocampo, A.; Tam, L.T.; Wood, L.S.; Pallegar, P.N.; Greene, J.J.; Geraghty, A.C.; Goldstein, A.K.; Ni, L.; et al. Methotrexate Chemotherapy Induces Persistent Tri-glial Dysregulation that Underlies Chemotherapy-Related Cognitive Impairment. Cell 2019, 176, 43–55.e13. [Google Scholar] [CrossRef] [Green Version]
- Da Mesquita, S.; Fu, Z.; Kipnis, J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron 2018, 100, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Pascual, O.; Ben, A.S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef] [Green Version]
- Pribiag, H.; Stellwagen, D. TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J. Neurosci. 2013, 33, 15879–15893. [Google Scholar] [CrossRef] [PubMed]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, V.H.; Anthony, D.C.; Bolton, S.J.; Brown, H.C. The blood-brain barrier and the inflammatory response. Mol. Med. Today 1997, 3, 335–341. [Google Scholar] [CrossRef]
- Anthony, D.C.; Bolton, S.J.; Fearn, S.; Perry, V.H. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain 1997, 120, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, S.J.; Anthony, D.C.; Perry, V.H. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience 1998, 86, 1245–1257. [Google Scholar] [CrossRef]
- Blond, D.; Campbell, S.J.; Butchart, A.G.; Perry, V.H.; Anthony, D.C. Differential induction of interleukin-1beta and tumour necrosis factor-alpha may account for specific patterns of leukocyte recruitment in the brain. Brain Res. 2002, 958, 89–99. [Google Scholar] [CrossRef]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Fransen, N.L.; Hsiao, C.C.; van der Poel, M.; Engelenburg, H.J.; Verdaasdonk, K.; Vincenten, M.C.J.; Remmerswaal, E.B.M.; Kuhlmann, T.; Mason, M.R.J.; Hamann, J.; et al. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 2020, 143, 1714–1730. [Google Scholar] [CrossRef]
- Fernández-López, D.; Faustino, J.; Daneman, R.; Zhou, L.; Lee, S.Y.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J. Neurosci. 2012, 32, 9588–9600. [Google Scholar] [CrossRef]
- Hou, J.; Yang, X.; Li, S.; Cheng, Z.; Wang, Y.; Zhao, J.; Zhang, C.; Li, Y.; Luo, M.; Ren, H.; et al. Accessing neuroinflammation sites: Monocyte/neutrophil-mediated drug delivery for cerebral ischemia. Sci. Adv. 2019, 5, eaau8301. [Google Scholar] [CrossRef] [Green Version]
- Tsao, N.; Hsu, H.P.; Wu, C.M.; Liu, C.C.; Lei, H.Y. Tumour necrosis factor-alpha causes an increase in blood-brain barrier permeability during sepsis. J. Med. Microbiol. 2001, 50, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Nishioku, T.; Dohgu, S.; Takata, F.; Eto, T.; Ishikawa, N.; Kodama, K.B.; Nakagawa, S.; Yamauchi, A.; Kataoka, Y. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol. Neurobiol. 2009, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.; Rogerson, S.; Taylor, T.; Tembo, M.; Mwenechanya, J.; Molyneux, M.; Turner, G. Blood-brain barrier function in cerebral malaria in Malawian children. Am. J. Trop. Med. Hyg. 2001, 64, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Brown, H.; Turner, G. Breaking down the blood-brain barrier: Signaling a path to cerebral malaria? Trends Parasitol. 2002, 18, 360–366. [Google Scholar] [CrossRef]
- Drevets, D.A.; Leenen, P.J.; Greenfield, R.A. Invasion of the central nervous system by intracellular bacteria. Clin. Microbiol. Rev. 2004, 17, 323–347. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.; Etienne-Manneville, S.; Adamson, P.; Couraud, P.O. Lymphocyte migration into the central nervous system: Implication of ICAM-1 signalling at the blood-brain barrier. Vascul. Pharmacol. 2002, 38, 315–322. [Google Scholar] [CrossRef]
- Mahajan, S.D.; Parikh, N.U.; Woodruff, T.M.; Jarvis, J.N.; Lopez, M.; Hennon, T.; Cunningham, P.; Quigg, R.J.; Schwartz, S.A.; Alexander, J.J. C5a alters blood-brain barrier integrity in a human In Vitro model of systemic lupus erythematosus. Immunology 2015, 146, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Boillat, M.; Hammoudi, P.M.; Dogga, S.K.; Pagès, S.; Goubran, M.; Rodriguez, I.; Soldati-Favre, D. Neuroinflammation-Associated Aspecific Manipulation of Mouse Predator Fear by Toxoplasma gondii. Cell Rep. 2020, 30, 320–334.e6. [Google Scholar] [CrossRef]
- Kano, S.I.; Hodgkinson, C.A.; Jones-Brando, L.; Eastwood, S.; Ishizuka, K.; Niwa, M.; Choi, E.Y.; Chang, D.J.; Chen, Y.; Velivela, S.D.; et al. Host-parasite interaction associated with major mental illness. Mol. Psychiatry 2020, 25, 194–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yolken, R.H.; Torrey, E.F. Viruses, schizophrenia, and bipolar disorder. Clin. Microbiol. Rev. 1995, 8, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Ackenheil, M. Psychoneuroimmunology and the cytokine action in the CNS: Implications for psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 1998, 22, 1–33. [Google Scholar] [CrossRef]
- Hanson, D.R.; Gottesman, I.I. Theories of schizophrenia: A genetic-inflammatory-vascular synthesis. BMC Med. Genet. 2005, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kety, S.S.; Woodford, R.B.; Harmel, M.H.; Freyman, F.A.; Appel, K.E.; Schmidt, C.F. Cerebral blood blow and metabolism in schizophrenia. Am. J. Psychiatry 1948, 104, 765–770. [Google Scholar] [CrossRef]
- Weinberger, D.R.; Berman, K.F. Speculation on the meaning of cerebral metabolic hypofrontality in schizophrenia. Schizophr. Bull. 1988, 14, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Ashton, L.; Barnes, A.; Livingston, M.; Wyper, D. Scottish Schizophrenia Research Group. Cingulate abnormalities associated with PANSS negative scores in first episode schizophrenia. Behav. Neurol. 2000, 12, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Franck, N.; O’Leary, D.S.; Flaum, M.; Hichwa, R.D.; Andreasen, N.C. Cerebral blood flow changes associated with Schneiderian first-rank symptoms in schizophrenia. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 277–282. [Google Scholar] [CrossRef]
- Paradiso, S.; Andreasen, N.C.; Crespo-Facorro, B.; O’Leary, D.S.; Watkins, G.L.; Boles, P.L.L.; Hichwa, R.D. Emotions in unmedicated patients with schizophrenia during evaluation with positron emission tomography. Am. J. Psychiatry 2003, 160, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Prata, J.; Santos, S.G.; Almeida, M.I.; Coelho, R.; Barbosa, M.A. Bridging Autism Spectrum Disorders and Schizophrenia through inflammation and biomarkers—pre-clinical and clinical investigations. J. Neuroinflamm. 2017, 14, 179. [Google Scholar] [CrossRef] [Green Version]
- Ting, E.Y.-C.; Yang, A.C.; Tsai, S.-J. Role of Interleukin-6 in Depressive Disorder. Int. J. Mol. Sci. 2020, 21, 2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Makinodan, M.; Toritsuka, M.; Okumura, K.; Kayashima, Y.; Ishida, R.; Kishimoto, N.; Takahashi, M.; Komori, T.; Yamaguchi, Y.; et al. TNF-α expression ration of M1/M2 macrophages is a potential adjunctive tool for the diagnosis of autism spectrum disorder. J. Neuroinflamm. 2020, in press. [Google Scholar]
- Onore, C.E.; Careaga, M.; Babineau, B.A.; Schwartzer, J.J.; Berman, R.F.; Ashwood, P. Inflammatory macrophage phenotype in BTBR T + tf/J mice. Front. Neurosci. 2013, 7, 158. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Meltzer, H.Y.; Bosmans, E.; Bergmans, R.; Vandoolaeghe, E.; Ranjan, R.; Desnyder, R. Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J. Affect. Disord. 1995, 34, 301–309. [Google Scholar] [CrossRef]
- Bai, Y.-M.; Chen, M.-H.; Hsu, J.-W.; Huang, K.-L.; Tu, P.-C.; Chang, W.-C.; Su, T.-P.; Li, C.T.; Lin, W.-C.; Tsai, S.-J. A comparison study of metabolic profiles, immunity, and brain gray matter volumes between patients with bipolar disorder and depressive disorder. J. Neuroinflamm. 2020, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, H.C.; Sullivan, E.L.; Battison, E.A.J.; Holton, K.F.; Graham, A.M.; Karalunas, S.L.; Fair, D.A.; Loftis, J.M.; Nigg, J.T. Evaluation of maternal inflammation as a marker of future offspring ADHD symptoms: A prospective investigation. Brain Behav. Immun. 2020, 89, 350–356. [Google Scholar] [CrossRef]
- Taoka, T.; Masutani, Y.; Kawai, H.; Nakane, T.; Matsuoka, K.; Yasuno, F.; Kishimoto, T.; Naganawa, S. Evaluation of glymphatic system activity with the diffusion MR technique: Diffusion tensor image analysis along the perivascular space (DTI-ALPS) in Alzheimer’s disease cases. Jpn. J. Radiol. 2017, 35, 172–178. [Google Scholar] [CrossRef]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Liu, Z.; Ren, W.; Jiang, W. Acute lipopolysaccharide exposure facilitates epileptiform activity via enhanced excitatory synaptic transmission and neuronal excitability in vitro. Neuropsychiatr. Dis. Treat. 2014, 10, 1489–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapal, L.; Igelhorst, B.A.; Dietzel-Meyer, I.D. Changes in Neuronal Excitability by Activated Microglia: Differential Na(+) Current Upregulation in Pyramid-Shaped and Bipolar Neurons by TNF-α and IL-18. Front. Neurol. 2016, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, G.; Inada, H.; Wake, H.; Akiyoshi, R.; Miyamoto, A.; Eto, K.; Ishikawa, T.; Moorhouse, A.J.; Strassman, A.M.; Nabekura, J. Microglial Contact Prevents Excess Depolarization and Rescues Neurons from Excitotoxicity. eNeuro 2016, 3, ENEURO.0004-16.2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segawa, K.; Blumenthal, Y.; Yamawaki, Y.; Ohtsuki, G. A Destruction Model of the Vascular and Lymphatic Systems in the Emergence of Psychiatric Symptoms. Biology 2021, 10, 34. https://doi.org/10.3390/biology10010034
Segawa K, Blumenthal Y, Yamawaki Y, Ohtsuki G. A Destruction Model of the Vascular and Lymphatic Systems in the Emergence of Psychiatric Symptoms. Biology. 2021; 10(1):34. https://doi.org/10.3390/biology10010034
Chicago/Turabian StyleSegawa, Kohei, Yukari Blumenthal, Yuki Yamawaki, and Gen Ohtsuki. 2021. "A Destruction Model of the Vascular and Lymphatic Systems in the Emergence of Psychiatric Symptoms" Biology 10, no. 1: 34. https://doi.org/10.3390/biology10010034
APA StyleSegawa, K., Blumenthal, Y., Yamawaki, Y., & Ohtsuki, G. (2021). A Destruction Model of the Vascular and Lymphatic Systems in the Emergence of Psychiatric Symptoms. Biology, 10(1), 34. https://doi.org/10.3390/biology10010034