Response of Human Glioblastoma Cells to Vitamin B12 Deficiency: A Study Using the Non-Toxic Cobalamin Antagonist

 , , , ,
, , , ,  , and
, and 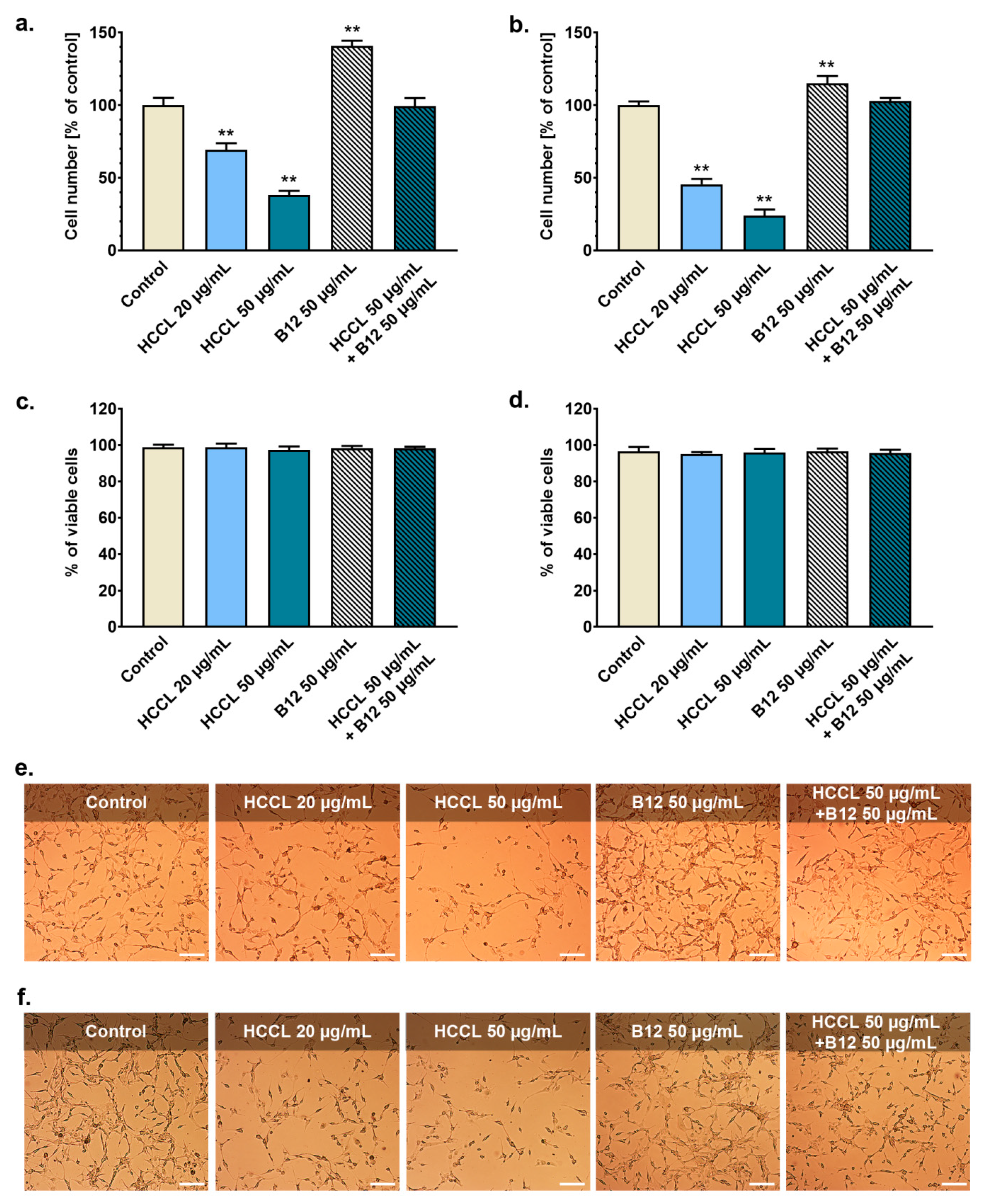{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Viability and Cell Count Assay
2.4. The Induction of Vitamin B12 Deficiency
2.5. Homocysteine Quantitative Analysis
2.6. Determination of Cell Cycle Distribution
2.7. In Vivo Toxicity
2.8. Molecular Docking
2.8.1. Macromolecule Preparation
2.8.2. Ligand Preparation
2.8.3. Docking of the Ligands
2.9. Statistics
3. Results
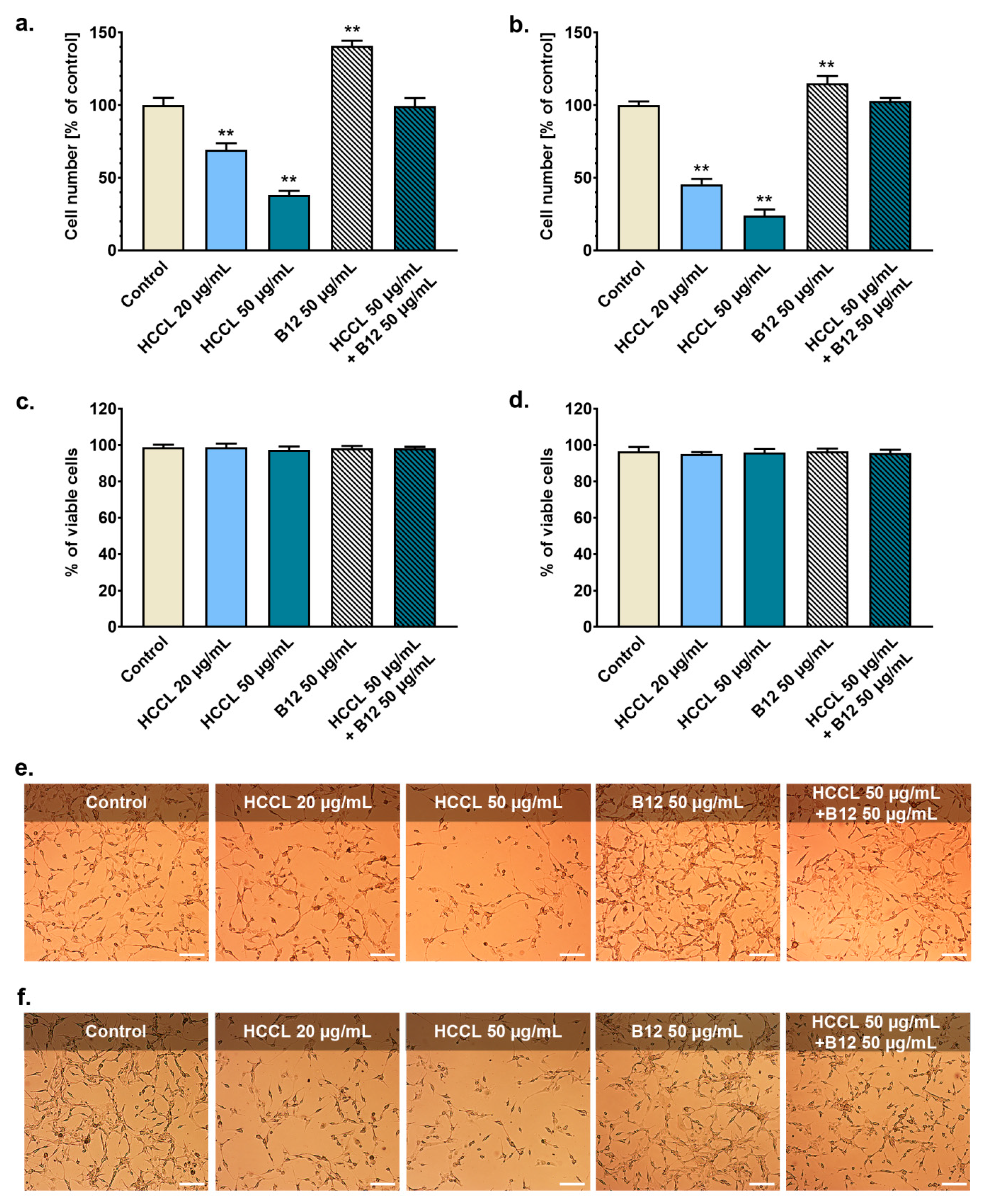
3.1. HCCL Inhibits Glioblastoma Cells Growth
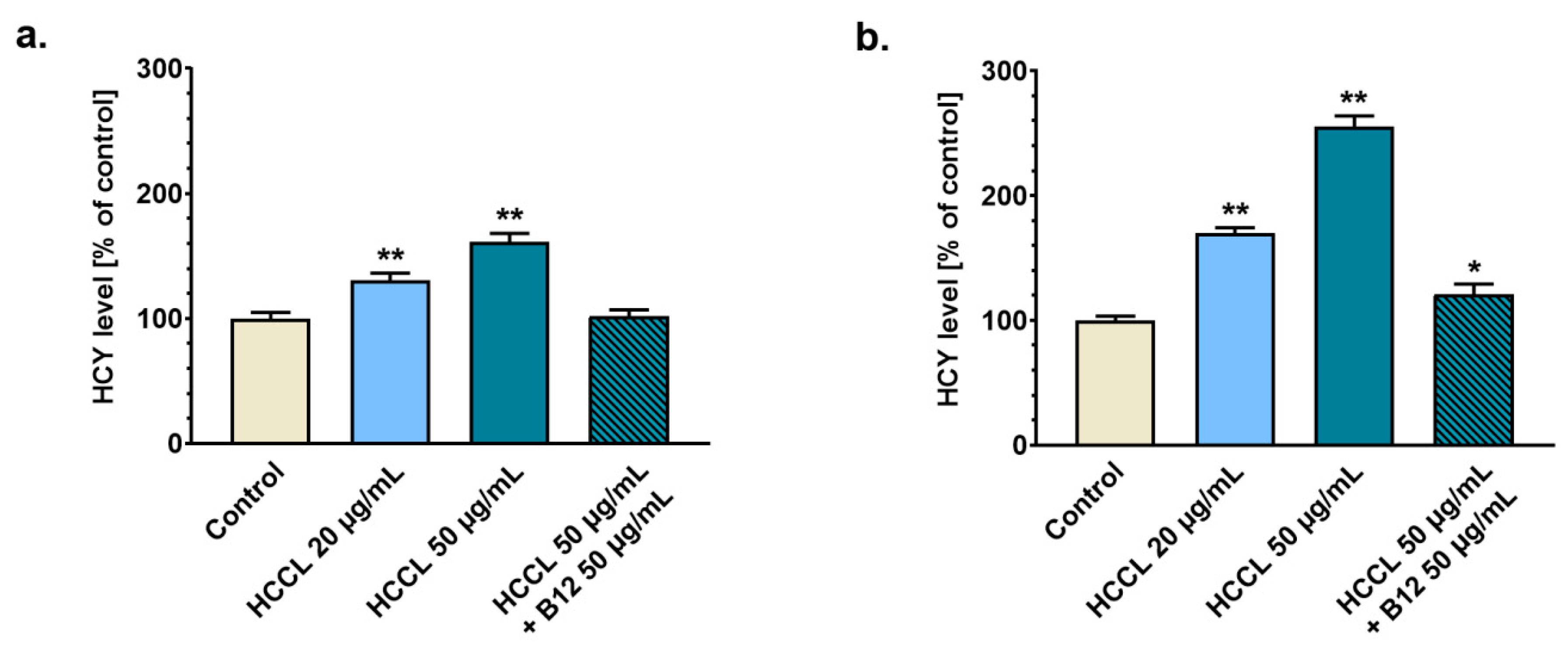
3.2. Treatment with HCCL Enhances Homocysteine Level in Glioblastoma Cell Culture
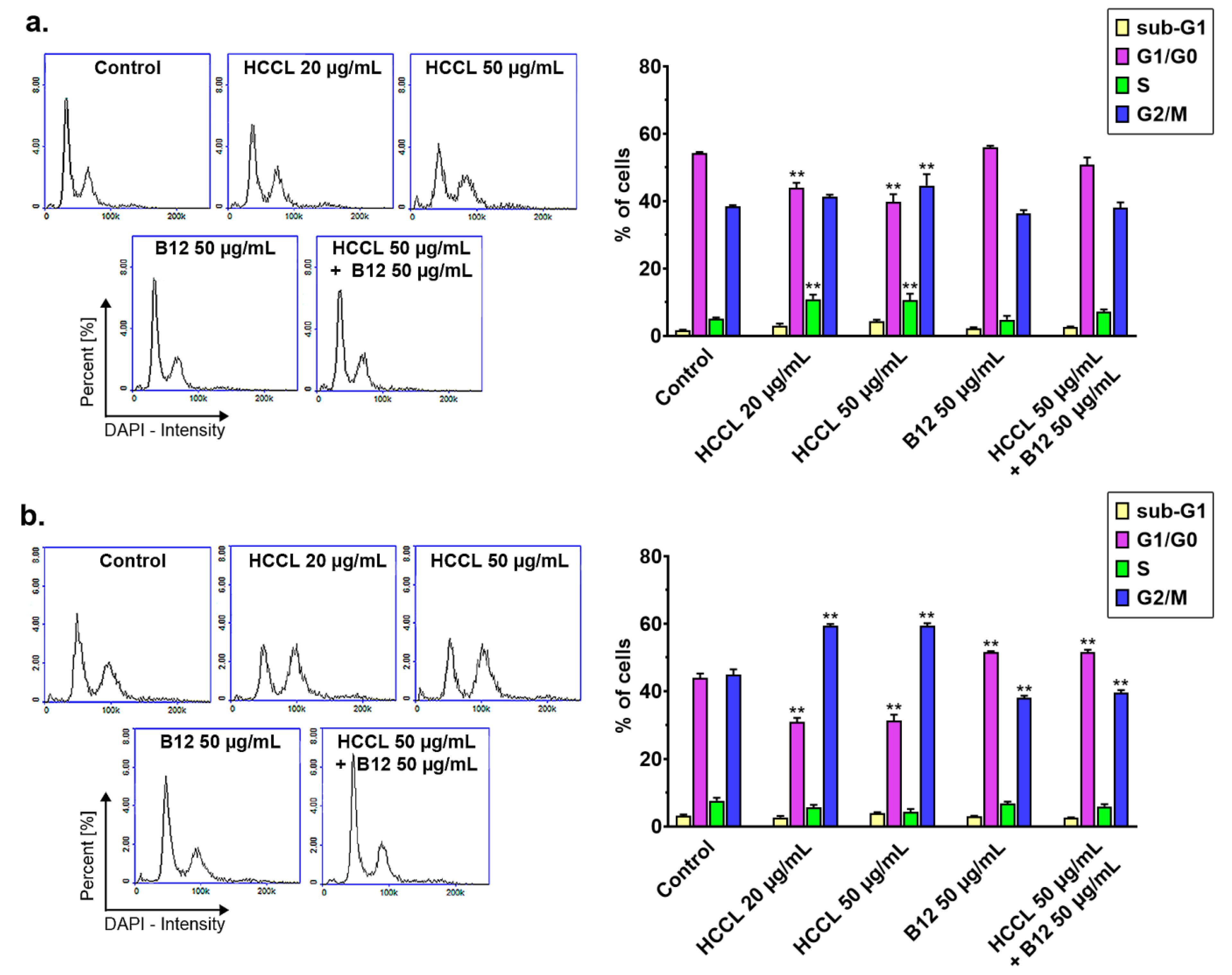
3.3. HCCL Disturbs Cell Cycle Progression of Glioblastoma Cells
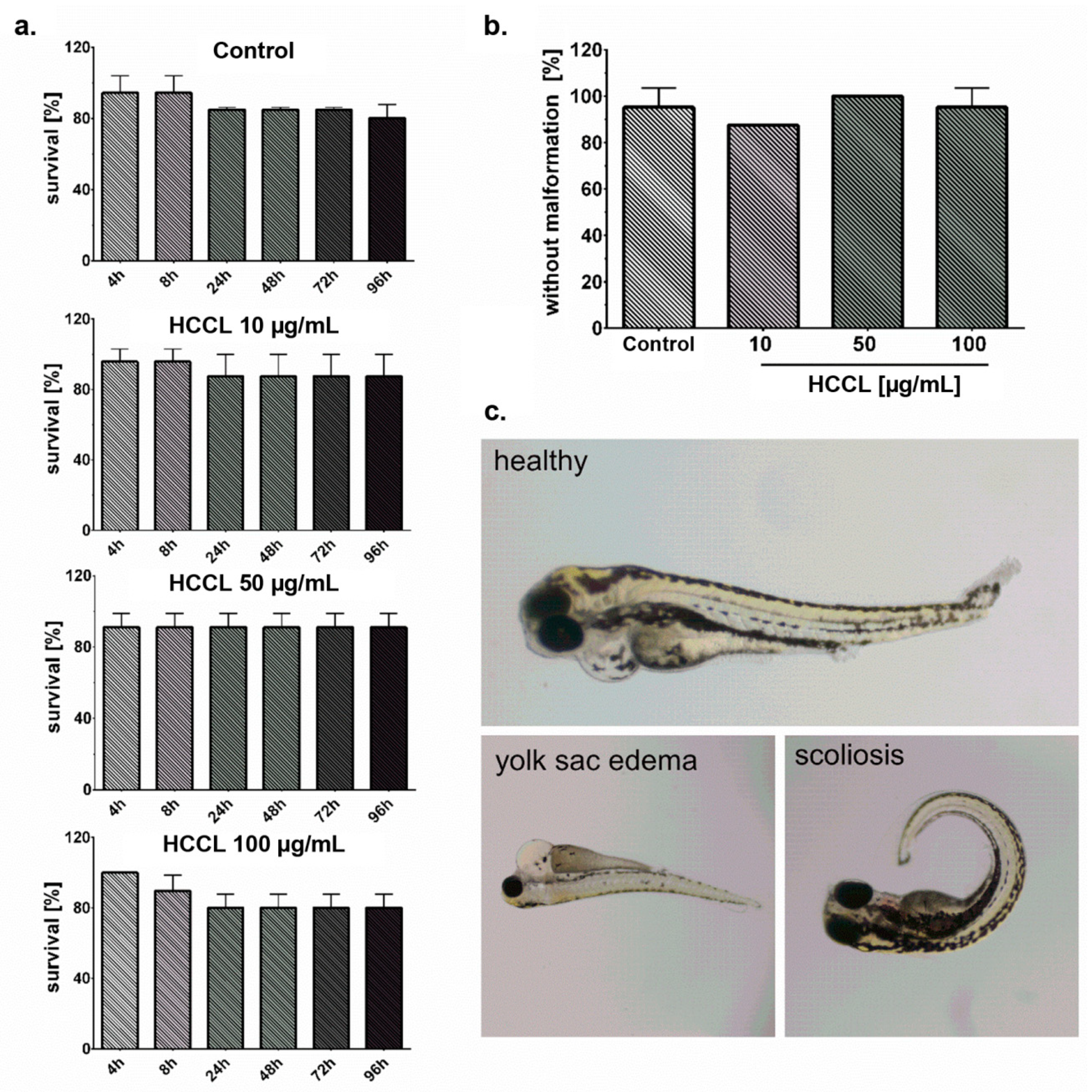
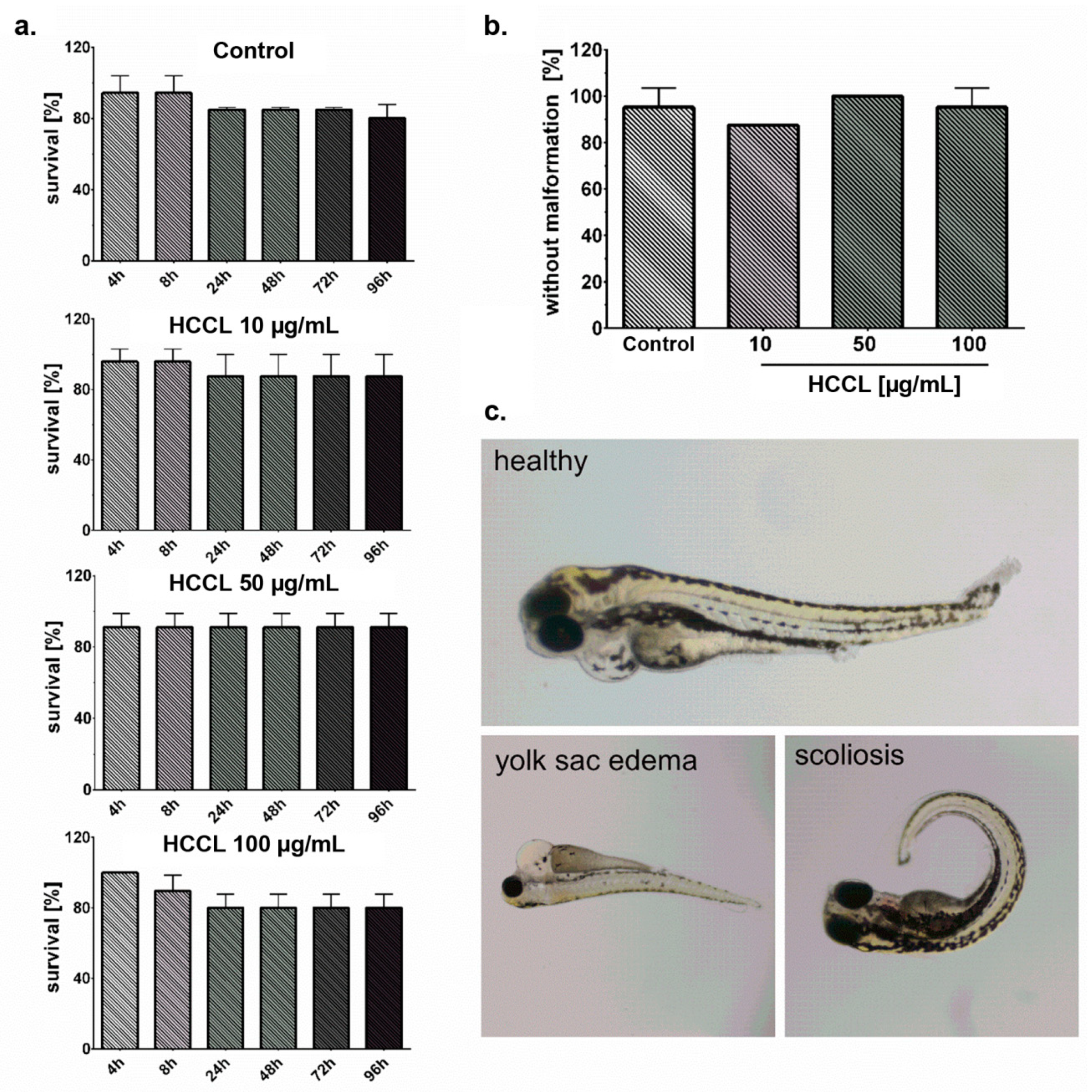
3.4. HCCL Does Not Induce Mortality or Malformations in Zebrafish Embryos
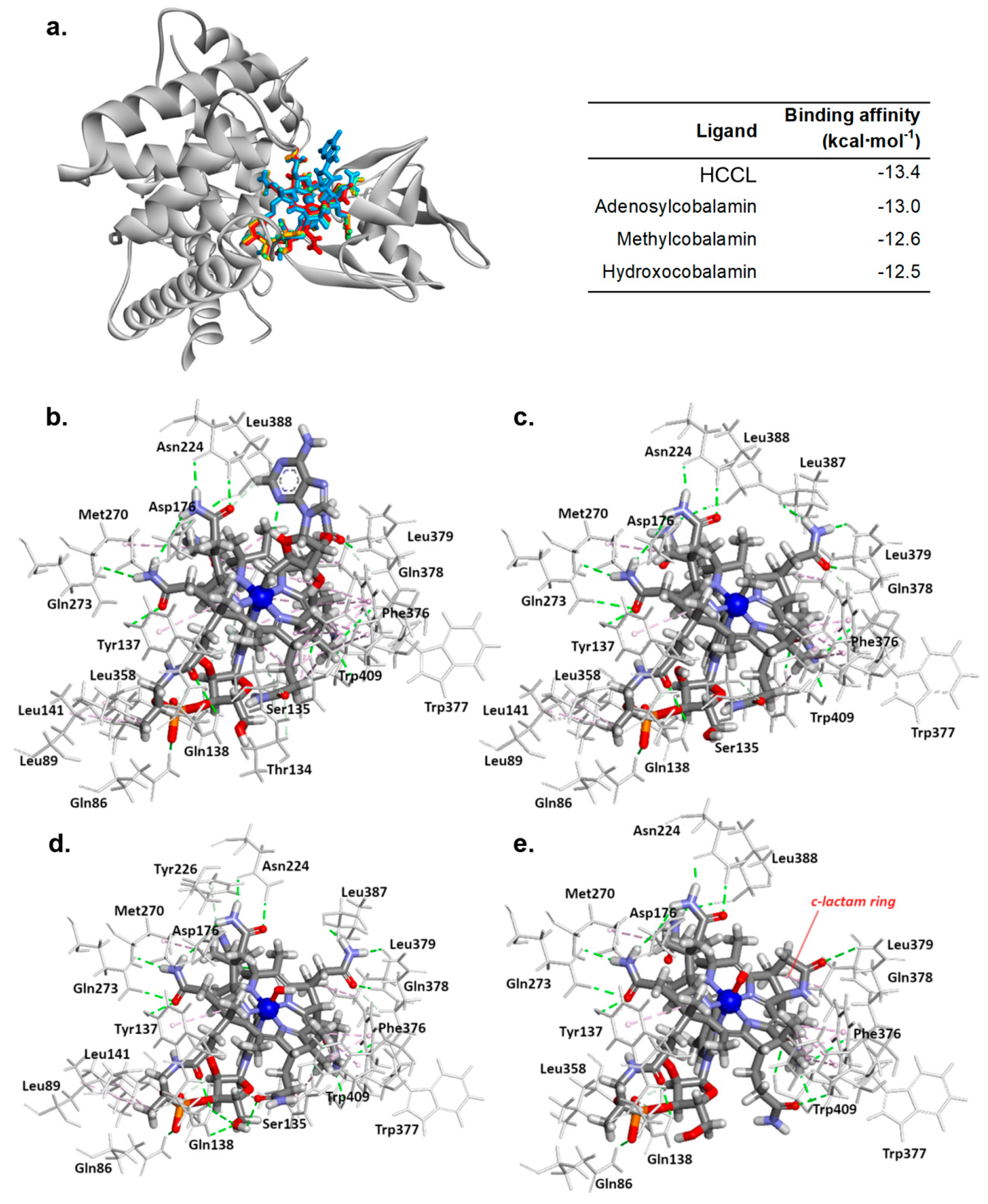
3.5. HCCL Binds to the Transcobalamin II Active Site as Natural Cobalamins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Alexander, B.M.; Cloughesy, T.F. Adult glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Batash, R.; Asna, N.; Schaffer, P.; Francis, N.; Schaffer, M. Glioblastoma multiforme, diagnosis and treatment; Recent Literature Review. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, R.K.; Parrish, K.E.; Sio, T.T.; Mittapalli, R.K.; Elmquist, W.F.; Sarkaria, J.N. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro-Oncology 2016, 18, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sysel, A.M.; Valli, V.E.; Nagle, R.B.; Bauer, J.A. Immunohistochemical quantification of the vitamin B12 transport protein (TCII), cell surface receptor (TCII-R) and Ki-67 in human tumor xenografts. Anticancer Res. 2013, 33, 4203–4212. [Google Scholar]
- Zelder, F.; Sonnay, M.; Prieto, L. Antivitamins for medicinal applications. Chembiochem 2015, 16, 1264–1278. [Google Scholar] [CrossRef]
- Shrier, M.S.; Trivedi, M.S.; Deth, R.C. Redox-Related epigenetic mechanisms in glioblastoma: Nuclear factor (erythroid-derived 2)-like 2, cobalamin, and dopamine receptor subtype 4. Front. Oncol. 2017, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrès, E.; Fothergill, H.; Mecili, M. Efficacy of oral cobalamin (vitamin B12) therapy. Expert Opin. Pharmacother. 2010, 11, 249–256. [Google Scholar] [CrossRef]
- Quadros, E.V.; Sequeira, J.M. Cellular uptake of cobalamin: Transcobalamin and the TCblR/CD320 receptor. Biochimie 2013, 95, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.L.; Brito, A.; Guéant, J.L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.H.; et al. Vitamin B12 deficiency. Nat. Rev. Dis. Primers 2017, 3, 17040. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Kamynina, E.; Field, M.S.; Stover, P.J. Folate rescues vitamin B12 depletion-induced inhibition of nuclear thymidylate biosynthesis and genome instability. Proc. Natl. Acad. Sci. USA 2017, 114, E4095–E4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brass, E.P.; Allen, R.H.; Ruff, L.J.; Stabler, S.P. Effect of hydroxycobalamin[c-lactam] on propionate and carnitine metabolism in the rat. Biochem. J. 1990, 266, 809–815. [Google Scholar]
- Stabler, S.P.; Brass, E.P.; Marcell, P.D.; Allen, R.H. Inhibition of cobalamin-dependent enzymes by cobalamin analogues in rats. J. Clin. Investig. 1991, 87, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Sponne, I.E.; Gaire, D.; Stabler, S.P.; Droesch, S.; Barbé, F.M.; Allen, R.H.; Lambert, D.A.; Nicolas, J.P. Inhibition of vitamin B12 metabolism by OH-cobalamin c-lactam in rat oligodendrocytes in culture: A model for studying neuropathy due to vitamin B12 deficiency. Neurosci. Lett. 2000, 288, 191–194. [Google Scholar] [CrossRef]
- Sauer, S.W.; Opp, S.; Haarmann, A.; Okun, J.G.; Kölker, S.; Morath, M.A. Long-term exposure of human proximal tubule cells to hydroxycobalamin[c-lactam] as a possible model to study renal disease in methylmalonic acidurias. J. Inherit. Metab. Dis. 2009, 32, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Haegler, P.; Grünig, D.; Berger, B.; Krähenbühl, S.; Bouitbir, J. Impaired mitochondrial function in HepG2 cells treated with hydroxy-cobalamin[c-lactam]: A cell model for idiosyncratic toxicity. Toxicology 2015, 336, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Haegler, P.; Grünig, D.; Berger, B.; Terracciano, L.; Krähenbühl, S.; Bouitbir, J. Hepatic effects of pharmacological doses of hydroxy-cobalamin[c-lactam] in mice. PLoS ONE 2017, 12, e0171026. [Google Scholar] [CrossRef]
- Rzepka, Z.; Respondek, M.; Beberok, A.; óProinsias, K.; Gryko, D.; Wrześniok, D. Vitamin B12 deficiency induces imbalance in melanocytes homeostasis—A cellular basis of hypocobalaminemia pigmentary manifestations. Int. J. Mol. Sci. 2018, 19, 2845. [Google Scholar] [CrossRef] [Green Version]
- Rzepka, Z.; Respondek, M.; Pawlik, J.; Beberok, A.; Gryko, D.; Wrześniok, D. Cobalamin deficiency: Effect on homeostasis of cultured human astrocytes. Cells 2019, 8, 1505. [Google Scholar] [CrossRef] [Green Version]
- Battaglia-Hsu, S.F.; Akchiche, N.; Noel, N.; Alberto, J.M.; Jeannesson, E.; Orozco-Barrios, C.E.; Martinez-Fong, D.; Daval, J.L.; Guéant, J.L. Vitamin B12 deficiency reduces proliferation and promotes differentiation of neuroblastoma cells and up-regulates PP2A, proNGF, and TACE. Proc. Natl. Acad. Sci. USA 2009, 106, 21930–21935. [Google Scholar] [CrossRef] [Green Version]
- Ghemrawi, R.; Pooya, S.; Lorentz, S.; Gauchotte, G.; Arnold, C.; Gueant, J.L.; Battaglia-Hsu, S.F. Decreased vitamin B12 availability induces ER stress through impaired SIRT1-deacetylation of HSF1. Cell Death Dis. 2013, 4, e553. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. Folate (vitamin B9) and vitamin B12 and their function in the maintenance of nuclear and mitochondrial genome integrity. Mutat. Res. 2012, 733, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, L.; Lysne, V.; Bjørke-Monsen, A.L.; Behringer, S.; Grünert, S.C.; Spiekerkoetter, U.; Jacobsen, D.W.; Blom, H.J. Biomarkers and algorithms for the diagnosis of vitamin B12 deficiency. Front. Mol. Biosci. 2016, 3, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultberg, B.; Andersson, A.; Isaksson, A. Metabolism of homocysteine, its relation to the other cellular thiols and its mechanism of cell damage in a cell culture line (human histiocytic cell line U-937). Biochim. Biophys. Acta 1995, 1269, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Darzynkiewicz, Z. Critical aspects in analysis of cellular DNA content. Curr. Protoc. Cytom. 2011. [Google Scholar] [CrossRef] [PubMed]
- Busquet, F.; Strecker, R.; Rawlings, J.M.; Belanger, S.E.; Braunbeck, T.; Carr, G.J.; Cenijn, P.; Fochtman, P.; Gourmelon, A.; Hübler, N.; et al. OECD validation study to assess intra- and inter-laboratory reproducibility of the zebrafish embryo toxicity test for acute aquatic toxicity testing. Regul. Toxicol. Pharmacol. 2014, 69, 496–511. [Google Scholar] [CrossRef]
- OECD. Test No. 236: Fish Embryo Acute Toxicity (FET) Test; OECD Publishing: Paris, France, 2013. [Google Scholar]
- Alam, A.; Woo, J.S.; Schmitz, J.; Printz, B.; Root, K.; Chen, F.; Bloch, J.S.; Zenobi, R.; Locher, K.P. Structural basis of transcobalamin recognition by human CD320 receptor. Nat. Commun. 2016, 7, 12100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.F. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchinson, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, C.; Brady, D.M. Comparative bioavailability and utilization of particular forms of B12 supplements with potential to mitigate B12-related genetic polymorphisms. Integr. Med. (Encinitas) 2017, 16, 42–49. [Google Scholar]
- Evans, V.J.; Bryant, J.C.; Mcquilkin, W.T.; Fioramonti, M.C.; Sanford, K.K.; Westfall, B.B.; Earle, W.R. Studies of nutrient media for tissue cells in vitro. II. An improved protein-free chemically defined medium for long-term cultivation of strain L-929 cells. Cancer Res. 1956, 16, 87–94. [Google Scholar]
- Amagasaki, T.; Green, R.; Jacobsen, D.W. Expression of transcobalamin II receptors by human leukemia K562 and HL-60 cells. Blood 1990, 76, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.X.; Poovey, C.E.; Privette, A.A.; Grant, G.D.; Chao, H.Y.; Cook, J.G.; Purvis, J.E. Orchestration of DNA damage checkpoint dynamics across the human cell cycle. Cell Syst. 2017, 5, 445–459.e5. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell. Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.; Rhind, N. Regulation of DNA replication by the S-phase DNA damage checkpoint. Cell Div. 2009, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Houtgraaf, J.H.; Versmissen, J.; van der Giessen, W.J. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc. Revasc. Med. 2006, 7, 165–172. [Google Scholar] [CrossRef]
- Chen, K.C.; Yang, T.Y.; Wu, C.C.; Cheng, C.C.; Hsu, S.L.; Hung, H.W.; Chen, J.W.; Chang, G.C. Pemetrexed induces S-phase arrest and apoptosis via a deregulated activation of Akt signaling pathway. PLoS ONE 2014, 9, e97888. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Yano, S. Tumor-specific S/G2-phase cell cycle arrest of cancer cells by methionine restriction. In Methionine Dependance of Cancer and Aging: Methods and Protocols, Methods in Molecular Biology, 1st ed.; Hoffman, R.M., Ed.; Springer: New York, NY, USA, 2019; Volume 1866, pp. 49–60. [Google Scholar] [CrossRef]
- Horzmann, K.A.; Freeman, J.L. Making Waves: New developments in toxicology with the zebrafish. Toxicol. Sci. 2018, 163, 5–12. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzepka, Z.; Rok, J.; Maszczyk, M.; Beberok, A.; Hermanowicz, J.M.; Pawlak, D.; Gryko, D.; Wrześniok, D. Response of Human Glioblastoma Cells to Vitamin B12 Deficiency: A Study Using the Non-Toxic Cobalamin Antagonist. Biology 2021, 10, 69. https://doi.org/10.3390/biology10010069
Rzepka Z, Rok J, Maszczyk M, Beberok A, Hermanowicz JM, Pawlak D, Gryko D, Wrześniok D. Response of Human Glioblastoma Cells to Vitamin B12 Deficiency: A Study Using the Non-Toxic Cobalamin Antagonist. Biology. 2021; 10(1):69. https://doi.org/10.3390/biology10010069
Chicago/Turabian StyleRzepka, Zuzanna, Jakub Rok, Mateusz Maszczyk, Artur Beberok, Justyna Magdalena Hermanowicz, Dariusz Pawlak, Dorota Gryko, and Dorota Wrześniok. 2021. "Response of Human Glioblastoma Cells to Vitamin B12 Deficiency: A Study Using the Non-Toxic Cobalamin Antagonist" Biology 10, no. 1: 69. https://doi.org/10.3390/biology10010069
APA StyleRzepka, Z., Rok, J., Maszczyk, M., Beberok, A., Hermanowicz, J. M., Pawlak, D., Gryko, D., & Wrześniok, D. (2021). Response of Human Glioblastoma Cells to Vitamin B12 Deficiency: A Study Using the Non-Toxic Cobalamin Antagonist. Biology, 10(1), 69. https://doi.org/10.3390/biology10010069