
Association between Sleep, Alzheimer’s, and Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. AD and Abnormal Proteins
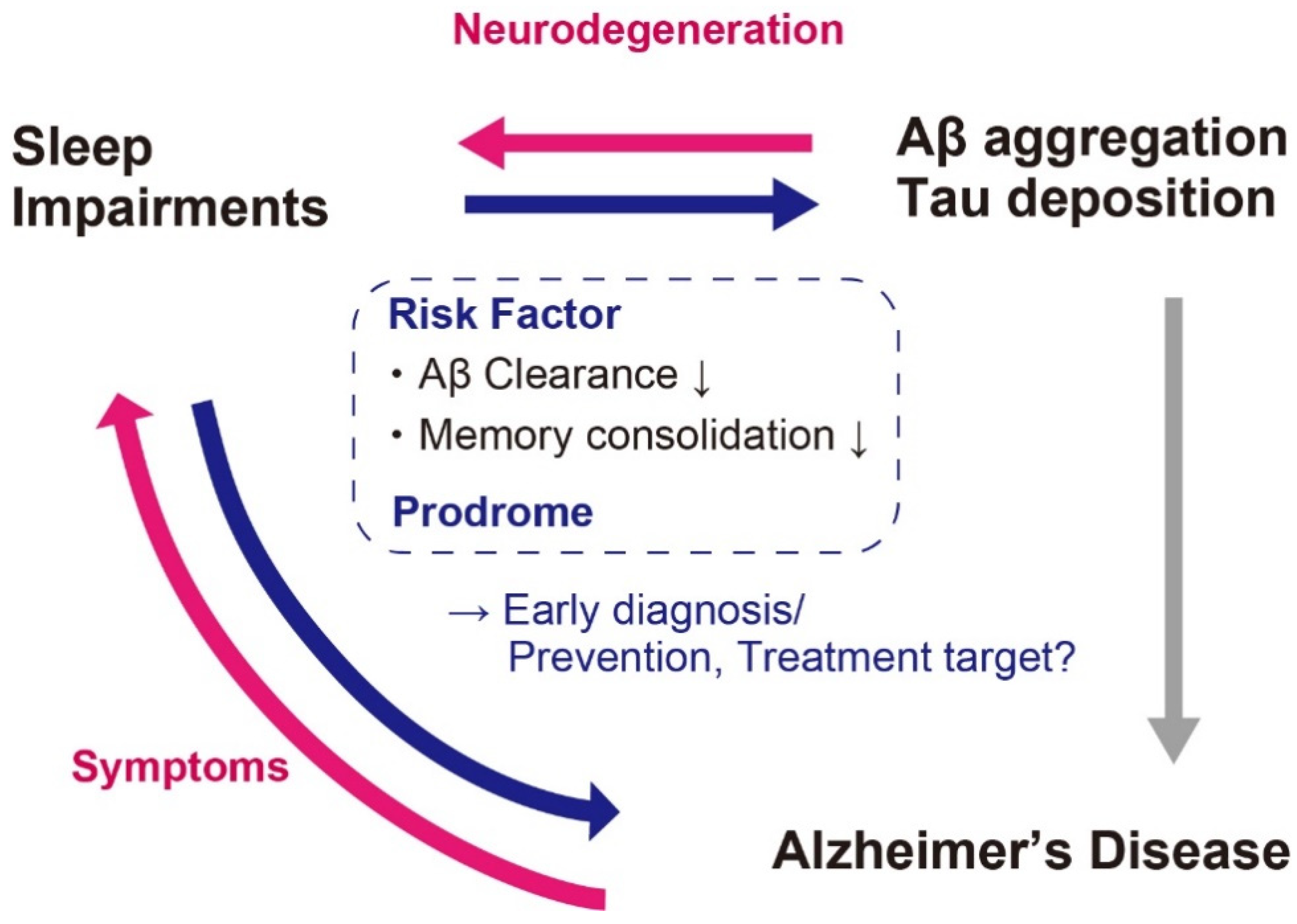
3. Aβ and Sleep Impairments in AD
4. Sleep Alterations Preceding Clinical Stages of AD
5. Prevention and Treatments of AD Targeting Sleep Impairments
6. PD and Abnormal Proteins
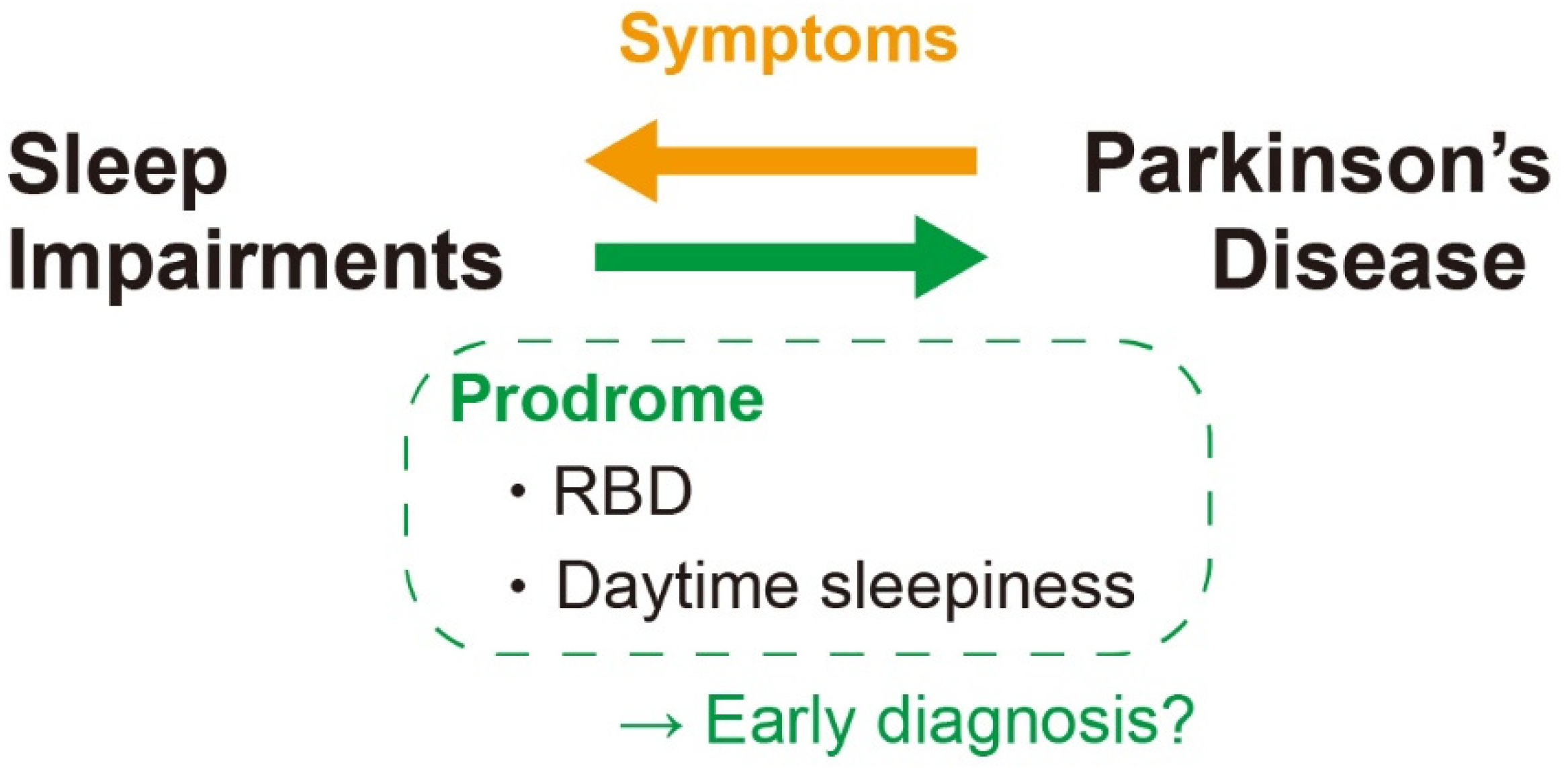
7. α-Syn and Sleep Impairments in PD
8. Sleep Impairments Preceding Clinical PD
9. Sleep Disorders as a Prevention and Treatment Target of PD
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, W. Economic Burden of Altzheimer Disease and Managed Care Considerations. Am. J. Manag. Care 2020, 26, S177–S183. [Google Scholar] [PubMed]
- Rizek, P.; Kumar, N.; Jog, M.S. An update on the diagnosis and treatment of Parkinson disease. CMAJ 2016, 188, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The Carboxy Terminus of the ß Amyloid Protein is Critical for the Seeding of β Amyloid Formation: Implications for the Pathogenesis of Alzheimer’s Disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Saito, T.; Suemoto, T.; Brouwers, N.; Sleegers, K.; Funamoto, S.; Mihira, N.; Matsuba, Y.; Yamada, K.; Nilsson, P.; Takano, J.; et al. Potent amyloidogenicity and pathogenicity of Aβ 243. Nat. Neurosci. 2011, 14, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Rodrigue, K.M.; Kennedy, K.M.; Devous, M.D.; Rieck, J.R.; Hebrank, A.C.; Diaz-Arrastia, R.; Mathews, D.; Park, D.C. β-Amyloid burden in healthy aging Regional distribution and cognitive consequences. Neurology 2012, 78, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccini, A.; Russo, C.; Gliozzi, A.; Relini, A.; Vitali, A.; Borghi, R.; Giliberto, L.; Armirotti, A.; D’Arrigo, C.; Bachi, A.; et al. β-amyloid is different in normal aging and in Alzheimer disease. J. Biol. Chem. 2005, 280, 34186–34192. [Google Scholar] [CrossRef] [Green Version]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Regan, P.; Whitcomb, D.J.; Cho, K. Physiological and pathophysiological implications of synaptic tau. Neuroscientist 2017, 23, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witman, G.B.; Cleveland, D.O.N.W.; Weingarten, M.D.; Kirschner, M.W. Tubulin requires tau for growth onto microtubule initiating sites. Proc. Natl. Acad. Sci. USA 1976, 73, 4070–4074. [Google Scholar] [CrossRef] [Green Version]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential Regulation of Dynein and Kinesin Motor Proteins by Tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.G.; Mirra, S.S.; Pollock, N.J.; Binder, L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (τ). Proc. Natl. Acad. Sci. USA 1986, 83, 4040–4043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, J.; Honda, T.; Mori, H.; Hamada, Y.; Miura, R.; Ogawara, M.; Ihara, Y. The carboxyl third of tau is tightly bound to paired helical filaments. Neuron 1988, 1, 827–834. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffrat, L.; Hayness, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid ~-protein similar to that in the senile plaques of Alzheimer1 s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer1 s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Venkitaramani, D.V.; Chin, J.; Netzer, W.J.; Gouras, G.K.; Lesne, S.; Malinow, R.; Lombroso, P.J. Β-Amyloid Modulation of Synaptic Transmission and Plasticity. J. Neurosci. 2007, 27, 11832–11837. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Kurup, P.; Zhang, Y.; Xu, J.; Venkitaramani, D.V.; Haroutunian, V.; Greengard, P.; Nairn, A.C.; Lombroso, P.J. Aβ-mediated NMDA receptor endocytosis in alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J. Neurosci. 2010, 30, 5948–5957. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Nairn, A.C.; Wang, P.; Lombroso, P.J. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 2003, 6, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR Removal Underlies Aβ-Induced Synaptic Depression and Dendritic Spine Loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.S.; Bloom, G.S. Tau: The Center of a Signaling Nexus in Alzheimer’s Disease. Front. Neurosci. 2016, 10, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martín, T.; Cuchillo-Ibáñez, I.; Noble, W.; Nyenya, F.; Anderton, B.H.; Hanger, D.P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 2013, 34, 2146–2157. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.A.; Lacroix, M.; Amar, F.; Larson, M.E.; Forster, C.; Aguzzi, A.; Bennett, D.A.; Ramsden, M.; Lesné, S.E. Soluble conformers of Aβ and tau alter selective proteins governing axonal transport. J. Neurosci. 2016, 36, 9647–9658. [Google Scholar] [CrossRef] [Green Version]
- Prinz, P.N.; Vitaliano, P.P.; Vitiello, M.V.; Bokan, J.; Raskind, M.; Peskind, E.; Gerber, C. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol. Aging 1982, 3, 361–370. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, B.; Adorni, F.; Musicco, M.; Appollonio, I.; Bonanni, E.; Caffarra, P.; Caltagirone, C.; Cerroni, G.; Concari, L.; Cosentino, F.I.I.; et al. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: A multicenter Italian clinical cross-sectional study on 431 patients. Dement. Geriatr. Cogn. Disord. 2012, 33, 50–58. [Google Scholar] [CrossRef] [Green Version]
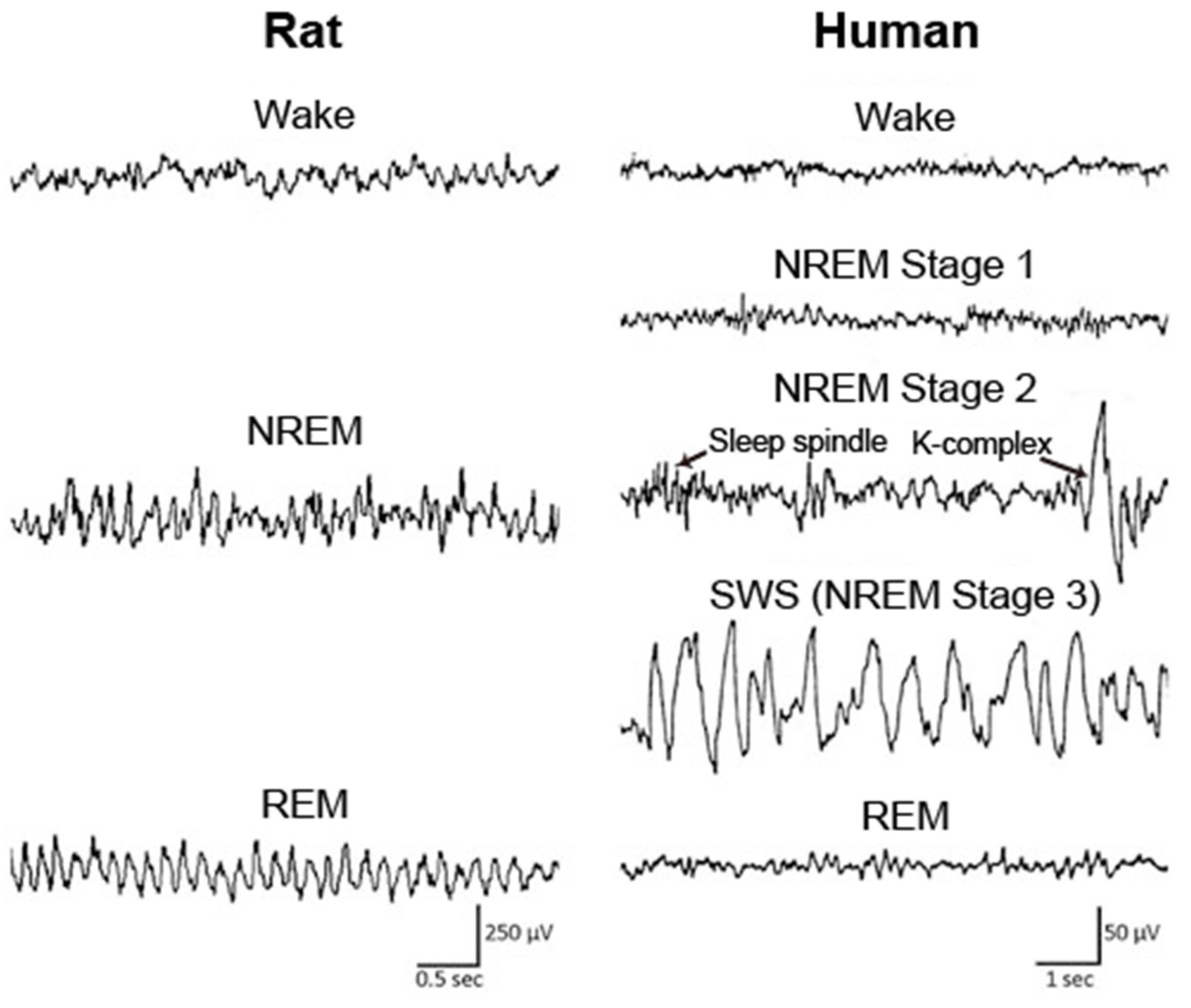
- McKenna, J.T.; Zielinski, M.R.; McCarley, R.W. Neurobiology of REM Sleep, NREM Sleep Homeostasis, and Gamma Band Oscillations. In Sleep Disorders Medicine; Springer: New York, NY, USA, 2017; pp. 55–77. ISBN 9781493965786. [Google Scholar]
- Landolt, H.P.; Dijk, D.J.; Achermann, P.; Borbély, A.A. Effect of age on the sleep EEG: Slow-wave activity and spindle frequency activity in young and middle-aged men. Brain Res. 1996, 738, 205–212. [Google Scholar] [CrossRef]
- Klerman, E.B.; Dijk, D.J. Age-Related Reduction in the Maximal Capacity for Sleep-Implications for Insomnia. Curr. Biol. 2008, 18, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Conte, F.; Arzilli, C.; Errico, B.M.; Giganti, F.; Iovino, D.; Ficca, G. Sleep Measures Expressing “Functional Uncertainty” in Elderlies’ Sleep. Gerontology 2014, 60, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Ohayon, M.M.; Carskadon, M.A.; Guilleminault, C.; Vitiello, M.V. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: Developing normative sleep values across the human lifespan. Sleep 2004, 27, 1255–1273. [Google Scholar] [CrossRef] [PubMed]
- Montplaisir, J.; Petit, D.; Lorrain, D.; Gauthier, S.; Nielsen, T. Sleep in Alzheimer’s disease: Further considerations on the role of brainstem and forebrain cholinergic populations in sleep-wake mechanisms. Sleep 1995, 18, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Tractenberg, R.E.; Singer, C.M.; Kaye, J.A. Characterizing sleep problems in persons with Alzheimer’s disease and normal elderly. J. Sleep Res. 2006, 15, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Ooms, S.; Ju, Y. El Treatment of Sleep Disorders in Dementia. Curr. Treat. Options Neurol. 2016, 18, 1–21. [Google Scholar] [CrossRef]
- Moran, M.; Lynch, C.A.; Walsh, C.; Coen, R.; Coakley, D.; Lawlor, B.A. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 2005, 6, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Mander, B.A.; Winer, J.R.; Jagust, W.J.; Walker, M.P. Sleep: A Novel Mechanistic Pathway, Biomarker, and Treatment Target in the Pathology of Alzheimer’s Disease? Trends Neurosci. 2016, 39, 552–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, D.; Gagnon, J.F.; Fantini, M.L.; Ferini-Strambi, L.; Montplaisir, J. Sleep and quantitative EEG in neurodegenerative disorders. J. Psychosom. Res. 2004, 56, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Kent, B.A.; Feldman, H.H.; Nygaard, H.B. Sleep and its regulation: An emerging pathogenic and treatment frontier in Alzheimer’s disease. Prog. Neurobiol. 2021, 197, 1019022. [Google Scholar] [CrossRef]
- Weber, F.; Dan, Y. Circuit-based interrogation of sleep control. Nature 2016, 538, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Sherin, J.E.; Shiromani, P.J.; McCarley, R.W.; Saper, C.B. Activation of ventrolateral preoptic neurons during sleep. Science 1996, 271, 216–219. [Google Scholar] [CrossRef] [Green Version]
- Sherin, J.E.; Elmquist, J.K.; Torrealba, F.; Saper, C.B. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J. Neurosci. 1998, 18, 4705–4721. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; McCormack, S.; España, R.A.; Crocker, A.; Scammell, T.E. Afferents to the orexin neurons of the rat brain. J. Comp. Neurol. 2006, 494, 845–861. [Google Scholar] [CrossRef] [Green Version]
- Steininger, T.L.; Gong, H.; Mcginty, D.; Szymusiak, R. Subregional organization of preoptic area/anterior hypothalamic projections to arousal-related monoaminergic cell groups. J. Comp. Neurol. 2001, 429, 638–653. [Google Scholar] [CrossRef]
- Lim, A.S.P.; Ellison, B.A.; Wang, J.L.; Yu, L.; Schneider, J.A.; Buchman, A.S.; Bennett, D.A.; Saper, C.B. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer’s disease. Brain 2014, 137, 2847–2861. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Greco, M.A.; Shiromani, P.; Saper, C.B. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J. Neurosci. 2000, 20, 3830–3842. [Google Scholar] [CrossRef] [Green Version]
- Teipel, S.J.; Flatz, W.H.; Heinsen, H.; Bokde, A.L.W.; Schoenberg, S.O.; Stöckel, S.; Dietrich, O.; Reiser, M.F.; Möller, H.J.; Hampel, H. Measurement of basal forebrain atrophy in Alzheimer’s disease using MRI. Brain 2005, 128, 2626–2644. [Google Scholar] [CrossRef] [Green Version]
- Teipel, S.; Heinsen, H.; Amaro, E.; Grinberg, L.T.; Krause, B.; Grothe, M. Cholinergic basal forebrain atrophy predicts amyloid burden in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P.; Maloney, A.J.F. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Cullen, K.M.; Halliday, G.M. Neurofibrillary degeneration and cell loss in the nucleus basalis in comparison to cortical Alzheimer pathology. Neurobiol. Aging 1998, 19, 297–306. [Google Scholar] [CrossRef]
- Mesulam, M.M.; Van Hoesen, G.W. Acetylcholinesterase-rich projections from the basal forebrain of the rhesus monkey to neocortex. Brain Res. 1976, 109, 152–157. [Google Scholar] [CrossRef]
- Xu, M.; Chung, S.; Zhang, S.; Zhong, P.; Ma, C.; Chang, W.C.; Weissbourd, B.; Sakai, N.; Luo, L.; Nishino, S.; et al. Basal forebrain circuit for sleep-wake control. Nat. Neurosci. 2015, 18, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Hassani, O.K.; Alonso, A.; Jones, B.E. Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J. Neurosci. 2005, 25, 4365–4369. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T. The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nat. Rev. Neurosci. 2007, 8, 171–181. [Google Scholar] [CrossRef]
- Fronczek, R.; van Geest, S.; Frölich, M.; Overeem, S.; Roelandse, F.W.C.; Lammers, G.J.; Swaab, D.F. Hypocretin (orexin) loss in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Buzsáki, G.; Anastassiou, C.A.; Koch, C. The origin of extracellular fields and currents-EEG, ECoG, LFP and spikes. Nat. Rev. Neurosci. 2012, 13, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Early network dysfunction in Alzheimer’s disease. Science 2019, 365, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Gerashchenko, D.; Timofeev, I.; Bacskai, B.J.; Kastanenka, K.V. Slow Wave Sleep Is a Promising Intervention Target for Alzheimer’s Disease. Front. Neurosci. 2020, 14, 1–11. [Google Scholar] [CrossRef]
- Soltani, S.; Chauvette, S.; Bukhtiyarova, O.; Lina, J.M.; Dubé, J.; Seigneur, J.; Carrier, J.; Timofeev, I. Sleep–Wake Cycle in Young and Older Mice. Front. Syst. Neurosci. 2019, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.H.; Huang, Y.; Bero, A.W.; Kasten, T.; Stewart, F.R.; Bateman, R.J.; Holtzman, D.M. Disruption of the sleep-wake cycle and diurnal fluctuation of amyloid-β in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 2012, 4, 150ra122. [Google Scholar] [CrossRef] [Green Version]
- Holth, J.K.; Mahan, T.E.; Robinson, G.O.; Rocha, A.; Holtzman, D.M. Altered sleep and EEG power in the P301S Tau transgenic mouse model. Ann. Clin. Transl. Neurol. 2017, 4, 180–190. [Google Scholar] [CrossRef]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [Green Version]
- Kastanenka, K.V.; Hou, S.S.; Shakerdge, N.; Logan, R.; Feng, D.; Wegmann, S.; Chopra, V.; Hawkes, J.M.; Chen, X.; Bacskai, B.J. Optogenetic restoration of disrupted slow oscillations halts amyloid deposition and restores calcium homeostasis in an animal model of Alzheimer’s disease. PLoS ONE 2017, 12, 1–25. [Google Scholar] [CrossRef]
- Yaffe, K.; Laffan, A.M.; Harrison, S.L.; Redline, S.; Spira, A.P.; Ensrud, K.E.; Ancoli-Israel, S.; Stone, K.L. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. Obstet. Gynecol. Surv. 2012, 67, 34–36. [Google Scholar] [CrossRef] [Green Version]
- Westerberg, C.E.; Mander, B.A.; Florczak, S.M.; Weintraub, S.; Mesulam, M.M.; Zee, P.C.; Paller, K.A. Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J. Int. Neuropsychol. Soc. 2012, 18, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.S.P.; Yu, L.; Kowgier, M.; Schneider, J.A.; Buchman, A.S.; Bennett, D.A. Modification of the relationship of the apolipoprotein E ε4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013, 70, 1544–1551. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.S.P.; Kowgier, M.; Yu, L.; Buchman, A.S.; Bennett, D.A. Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 2013, 36, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadotani, H.; Young, T.; Peppard, P.E.; Finn, L.; Colrain, I.M.; Murphy, G.M.; Page, P. Association Between Apolipoprotein E ϵ4. JAMA 2001, 285, 2888–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.E.S.; McLeland, J.S.; Toedebusch, C.D.; Xiong, C.; Fagan, A.M.; Duntley, S.P.; Morris, J.C.; Holtzman, D.M. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013, 70, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.P.; Gamaldo, A.A.; An, Y.; Wu, M.N.; Simonsick, E.M.; Bilgel, M.; Zhou, Y.; Wong, D.F.; Ferrucci, L.; Resnick, S.M. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013, 70, 1537–1543. [Google Scholar] [CrossRef] [Green Version]
- Mander, B.A.; Marks, S.M.; Vogel, J.W.; Rao, V.; Lu, B.; Saletin, J.M.; Ancoli-Israel, S.; Jagust, W.J.; Walker, M.P. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci. 2015, 18, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Mesulam, M.; Shaw, P.; Mash, D.; Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 2004, 55, 815–828. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Feld, G.B.; Born, J. Sculpting memory during sleep: Concurrent consolidation and forgetting. Curr. Opin. Neurobiol. 2017, 44, 20–27. [Google Scholar] [CrossRef]
- Raven, F.; Van der Zee, E.A.; Meerlo, P.; Havekes, R. The role of sleep in regulating structural plasticity and synaptic strength: Implications for memory and cognitive function. Sleep Med. Rev. 2018, 39, 3–11. [Google Scholar] [CrossRef]
- Ooms, S.; Overeem, S.; Besse, K.; Rikkert, M.O.; Verbeek, M.; Claassen, J.A.H.R. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men a randomized clinical trial. JAMA Neurol. 2014, 71, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Lucey, B.P.; Hicks, T.J.; McLeland, J.S.; Toedebusch, C.D.; Boyd, J.; Elbert, D.L.; Patterson, B.W.; Baty, J.; Morris, J.C.; Ovod, V.; et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 2018, 83, 197–204. [Google Scholar] [CrossRef]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- Shokri-Kojori, E.; Wang, G.J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Pizzo, M.E.; Preston, J.E.; Janigro, D.; Thorne, R.G. The role of brain barriers in fluid movement in the CNS: Is there a ‘glymphatic’ system? Acta Neuropathol. 2018, 135, 387–407. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.; Mlynczak, S.; Erickson, T.; Man, S.F.P.; Man, G.C.W. Oxygen consumption during sleep: Influence of sleep stage and time of night. Sleep 1989, 12, 201–210. [Google Scholar]
- DiNuzzo, M.; Nedergaard, M. Brain energetics during the sleep–wake cycle. Curr. Opin. Neurobiol. 2017, 47, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everson, C.A.; Henchen, C.J.; Szabo, A.; Hogg, N. Cell injury and repair resulting from sleep loss and sleep recovery in laboratory rats. Sleep 2014, 37, 1929–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villafuerte, G.; Miguel-Puga, A.; Murillo Rodríguez, E.; Machado, S.; Manjarrez, E.; Arias-Carrión, O. Sleep deprivation and oxidative stress in animal models: A systematic review. Oxid. Med. Cell. Longev. 2015, 2015, 234952. [Google Scholar] [CrossRef] [PubMed]
- Misonou, H.; Morishima-Kawashima, M.; Ihara, Y. Oxidative stress induces intracellular accumulation of amyloid β- protein (Aβ) in human neuroblastoma cells. Biochemistry 2000, 39, 6951–6959. [Google Scholar] [CrossRef]
- Yatin, S.M.; Varadarajan, S.; Link, C.D.; Butterfield, D.A. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid β-peptide (1-42). Neurobiol. Aging 1999, 20, 325–330. [Google Scholar]
- Dos Santos Moraes, W.A.; Poyares, D.R.; Guilleminault, C.; Ramos, L.R.; Ferreira Bertolucci, P.H.; Tufik, S. The effect of donepezil on sleep and REM sleep EEG in patients with Alzheimer disease: A double-blind placebo-controlled study. Sleep 2006, 29, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Moraes, W.; Poyares, D.; Sukys-Claudino, L.; Guilleminault, C.; Tufik, S. Donepezil improves obstructive sleep apnea in Alzheimer disease: A double-blind, placebo-controlled study. Chest 2008, 133, 677–683. [Google Scholar] [CrossRef]
- Roh, J.H.; Jiang, H.; Finn, M.B.; Stewart, F.R.; Mahan, T.E.; Cirrito, J.R.; Heda, A.; Joy Snider, B.; Li, M.; Yanagisawa, M.; et al. Correction to Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J. Exp. Med. 2014, 211, 2487–2496. [Google Scholar] [CrossRef]
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Orexin and the Sleep-Wake Cycle. Science 2009, 326, 1005–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beracochea, D. Anterograde and retrograde effects of benzodiazepines on memory. Sci. World J. 2006, 6, 1460–1465. [Google Scholar] [CrossRef]
- Shih, H.I.; Lin, C.C.; Tu, Y.F.; Chang, C.M.; Hsu, H.C.; Chi, C.H.; Kao, C.H. An increased risk of reversible dementia may occur after zolpidem derivative use in the elderly population a population-based case-control study. Medicine 2015, 94, e809. [Google Scholar] [CrossRef]
- Papalambros, N.A.; Santostasi, G.; Malkani, R.G.; Braun, R.; Weintraub, S.; Paller, K.A.; Zee, P.C. Acoustic enhancement of sleep slow oscillations and concomitant memory improvement in older adults. Front. Hum. Neurosci. 2017, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papalambros, N.A.; Weintraub, S.; Chen, T.; Grimaldi, D.; Santostasi, G.; Paller, K.A.; Zee, P.C.; Malkani, R.G. Acoustic enhancement of sleep slow oscillations in mild cognitive impairment. Ann. Clin. Transl. Neurol. 2019, 6, 1191–1201. [Google Scholar] [CrossRef] [Green Version]
- Ngo, H.V.V.; Martinetz, T.; Born, J.; Mölle, M. Auditory closed-loop stimulation of the sleep slow oscillation enhances memory. Neuron 2013, 78, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Besedovsky, L.; Ngo, H.V.V.; Dimitrov, S.; Gassenmaier, C.; Lehmann, R.; Born, J. Auditory closed-loop stimulation of EEG slow oscillations strengthens sleep and signs of its immune-supportive function. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Bayer, L.; Constantinescu, I.; Perrig, S.; Vienne, J.; Vidal, P.P.; Mühlethaler, M.; Schwartz, S. Rocking synchronizes brain waves during a short nap. Curr. Biol. 2011, 21, R461–R462. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Kuramoto, A.; Inui, Y.; Inou, N. Mechanical Bed for Investigating Sleep-Inducing Vibration. J. Healthc. Eng. 2017, 2017, 2364659. [Google Scholar] [CrossRef] [PubMed]
- Perrault, A.A.; Khani, A.; Quairiaux, C.; Kompotis, K.; Franken, P.; Muhlethaler, M.; Schwartz, S.; Bayer, L. Whole-Night Continuous Rocking Entrains Spontaneous Neural Oscillations with Benefits for Sleep and Memory. Curr. Biol. 2019, 29, 402–411.e3. [Google Scholar] [CrossRef] [Green Version]
- Kompotis, K.; Hubbard, J.; Emmenegger, Y.; Perrault, A.; Mühlethaler, M.; Schwartz, S.; Bayer, L.; Franken, P. Rocking Promotes Sleep in Mice through Rhythmic Stimulation of the Vestibular System. Curr. Biol. 2019, 29, 392–401.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s Disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Pacelli, C.; Giguère, N.; Bourque, M.J.; Lévesque, M.; Slack, R.S.; Trudeau, L.É. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8, 451–465. [Google Scholar] [CrossRef]
- Nath, S.; Goodwin, J.; Engelborghs, Y.; Pountney, D.L. Raised calcium promotes α-synuclein aggregate formation. Mol. Cell. Neurosci. 2011, 46, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur. J. Neurosci. 2006, 24, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.K.; Choi, M.G.; Kim, J.Y.; Yang, Y.; Lai, Y.; Kweon, D.H.; Lee, N.K.; Shin, Y.K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef] [Green Version]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Björklund, A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar] [CrossRef] [Green Version]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Taoufiq, Z.; Thorn-Seshold, O.; Trauner, D.; Hasegawa, M.; Takahashi, T. Wild-type monomeric α-synuclein can impair vesicle endocytosis and synaptic fidelity via tubulin polymerization at the calyx of held. J. Neurosci. 2017, 37, 6043–6052. [Google Scholar] [CrossRef]
- Xu, J.; Wu, X.S.; Sheng, J.; Zhang, Z.; Yue, H.Y.; Sun, L.; Sgobio, C.; Lin, X.; Peng, S.; Jin, Y.; et al. α-Synuclein mutation inhibits endocytosis at mammalian central nerve terminals. J. Neurosci. 2016, 36, 4408–4414. [Google Scholar] [CrossRef]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef]
- Suzuki, K.; Miyamoto, M.; Miyamoto, T.; Hirata, K. Parkinson’s Disease and Sleep/Wake Disturbances. Curr. Neurol. Neurosci. Rep. 2015, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Kovalzon, V.M.; Zavalko, I.M. The neurochemistry of the sleep-wakefulness cycle and Parkinson’s disease. Neurochem. J. 2013, 7, 171–183. [Google Scholar] [CrossRef]
- Kumar, S.; Bhatia, M.; Behari, M. Sleep disorders in Parkinson’s disease. Mov. Disord. 2002, 17, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Sixel-Döring, F.; Schweitzer, M.; Mollenhauer, B.; Trenkwalder, C. Intraindividual variability of REM sleep behavior disorder in Parkinson’s disease: A comparative assessment using a new REM sleep behavior disorder severity scale (RBDSS) for clinical routine. J. Clin. Sleep Med. 2011, 7, 75–80. [Google Scholar] [CrossRef]
- Keir, L.H.M.; Breen, D.P. New awakenings: Current understanding of sleep dysfunction and its treatment in Parkinson ’ s disease. J. Neurol. 2020, 267, 288–294. [Google Scholar] [CrossRef] [Green Version]
- French, I.T.; Muthusamy, K.A. A review of sleep and its disorders in patients with Parkinson’s disease in relation to various brain structures. Front. Aging Neurosci. 2016, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Héricé, C.; Patel, A.A.; Sakata, S. Circuit mechanisms and computational models of REM sleep. Neurosci. Res. 2019, 140, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, T.C.; Lai, Y.Y.; Siegel, J.M. Hypocretin (orexin) cell loss in Parkinson’s disease. Brain 2007, 130, 1586–1595. [Google Scholar] [CrossRef] [Green Version]
- Wienecke, M.; Werth, E.; Poryazova, R.; Baumann-Vogel, H.; Bassetti, C.L.; Weller, M.; Waldvogel, D.; Storch, A.; Baumann, C.R. Progressive dopamine and hypocretin deficiencies in Parkinson’s disease: Is there an impact on sleep and wakefulness? J. Sleep Res. 2012, 21, 710–717. [Google Scholar] [CrossRef]
- Ylikoski, A.; Martikainen, K.; Sarkanen, T.; Partinen, M. Parkinson’s disease and narcolepsy-like symptoms. Sleep Med. 2015, 16, 540–544. [Google Scholar] [CrossRef]
- Takahashi, J.S. Molecular neurobiology and genetics of circadian rhythms. Annu. Rev. Neurosci. 1995, 18, 531–553. [Google Scholar] [CrossRef]
- Harnois, C.; Di Paolo, T. Decreased dopamine in the retinas of patients with Parkinson’s disease. Investig. Ophthalmol. Vis. Sci. 1990, 31, 2473–2475. [Google Scholar]
- Videnovic, A.; Noble, C.; Reid, K.J.; Peng, J.; Turek, F.W.; Marconi, A.; Rademaker, A.W.; Simuni, T.; Zadikoff, C.; Zee, P.C. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol. 2014, 71, 463–469. [Google Scholar] [CrossRef]
- Sohail, S.; Yu, L.; Schneider, J.A.; Bennett, D.A.; Buchman, A.S.; Lim, A.S.P. Sleep fragmentation and Parkinson’s disease pathology in older adults without Parkinson’s disease. Mov. Disord. 2017, 32, 1729–1737. [Google Scholar] [CrossRef]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Högl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 2019, 142, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Lysen, T.S.; Darweesh, S.K.L.; Kamran Ikram, M.; Luik, A.I.; Arfan Ikram, M. Sleep and risk of parkinsonism and Parkinson’s disease: A population-based study. Brain 2019, 142, 2013–2022. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Huang, X.; Park, Y.; Hollenbeck, A.; Blair, A.; Schatzkin, A.; Chen, H. Daytime napping, nighttime sleeping, and parkinson disease. Am. J. Epidemiol. 2011, 173, 1032–1038. [Google Scholar] [CrossRef]
- Barraud, Q.; Lambrecq, V.; Forni, C.; McGuire, S.; Hill, M.; Bioulac, B.; Balzamo, E.; Bezard, E.; Tison, F.; Ghorayeb, I. Sleep disorders in Parkinson’s disease: The contribution of the MPTP non-human primate model. Exp. Neurol. 2009, 219, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Ikuno, M.; Hondo, M.; Parajuli, L.K.; Taguchi, K.; Ueda, J.; Sawamura, M.; Okuda, S.; Nakanishi, E.; Hara, J.; et al. α-synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: A prodromal Parkinson’s disease model. Brain 2020, 143, 249–265. [Google Scholar] [CrossRef]
- Shen, Y.; Yu, W.B.; Shen, B.; Dong, H.; Zhao, J.; Tang, Y.L.; Fan, Y.; Yang, Y.F.; Sun, Y.M.; Luo, S.S.; et al. Propagated a-synucleinopathy recapitulates REM sleep behaviour disorder followed by parkinsonian phenotypes in mice. Brain 2020, 143, 3374–3392. [Google Scholar] [CrossRef] [PubMed]
- Pastukhov, Y.F.; Simonova, V.V.; Chernyshev, M.V.; Guzeev, M.A.; Shemyakova, T.S.; Ekimova, I.V. Signs of sleep and behavior disorders indicating the initial stage of neurodegeneration in a rat model of Parkinson’s disease. J. Evol. Biochem. Physiol. 2017, 53, 431–434. [Google Scholar] [CrossRef]
- Boeve, B.F.; Silber, M.H.; Ferman, T.J. Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: Results in 14 patients. Sleep Med. 2003, 4, 281–284. [Google Scholar] [CrossRef]
- Dowling, G.A.; Mastick, J.; Colling, E.; Carter, J.H.; Singer, C.M.; Aminoff, M.J. Melatonin for sleep disturbances in Parkinson’s disease. Sleep Med. 2005, 6, 459–466. [Google Scholar] [CrossRef]
- Medeiros, C.A.M.; Carvalhedo De Bruin, P.F.; Lopes, L.A.; Magalhães, M.C.; De Lourdes Seabra, M.; Sales De Bruin, V.M. Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson’s disease: A randomized, double blind, placebo-controlled study. J. Neurol. 2007, 254, 459–464. [Google Scholar] [CrossRef]
- Liu, M.F.; Xue, Y.; Liu, C.; Liu, Y.H.; Diao, H.L.; Wang, Y.; Pan, Y.P.; Chen, L. Orexin—A exerts neuroprotective effects via OX1R in Parkinson⇔s disease. Front. Neurosci. 2018, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hadadianpour, Z.; Fatehi, F.; Ayoobi, F.; Kaeidi, A.; Shamsizadeh, A.; Fatemi, I. The effect of orexin-A on motor and cognitive functions in a rat model of Parkinson’s disease. Neurol. Res. 2017, 39, 845–851. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, S.; Tsunematsu, T. Association between Sleep, Alzheimer’s, and Parkinson’s Disease. Biology 2021, 10, 1127. https://doi.org/10.3390/biology10111127
Matsumoto S, Tsunematsu T. Association between Sleep, Alzheimer’s, and Parkinson’s Disease. Biology. 2021; 10(11):1127. https://doi.org/10.3390/biology10111127
Chicago/Turabian StyleMatsumoto, Sumire, and Tomomi Tsunematsu. 2021. "Association between Sleep, Alzheimer’s, and Parkinson’s Disease" Biology 10, no. 11: 1127. https://doi.org/10.3390/biology10111127
APA StyleMatsumoto, S., & Tsunematsu, T. (2021). Association between Sleep, Alzheimer’s, and Parkinson’s Disease. Biology, 10(11), 1127. https://doi.org/10.3390/biology10111127