
Siglecs as Therapeutic Targets in Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Siglec Family
2.1. Siglec-1 (CD169)
2.2. Siglec-2 (CD22)
2.3. Siglec-3 (CD33)
2.4. Siglec-4 (Myelin Associated Glycoprotein, MAG)
2.5. Siglec-5 (CD170) and Siglec-14 Pairs
2.6. Siglec-6 (CD327)
2.7. Siglec-7 (p75/AIRM1, CD328)
2.8. Siglec-8
2.9. Siglec-9 (CD329)
2.10. Siglec-10
2.11. Siglec-11 and Siglec-16 Pairs
2.12. Siglec-12 (Siglec-XII)
2.13. Siglec-13
2.14. Siglec-15
2.15. Siglec-17
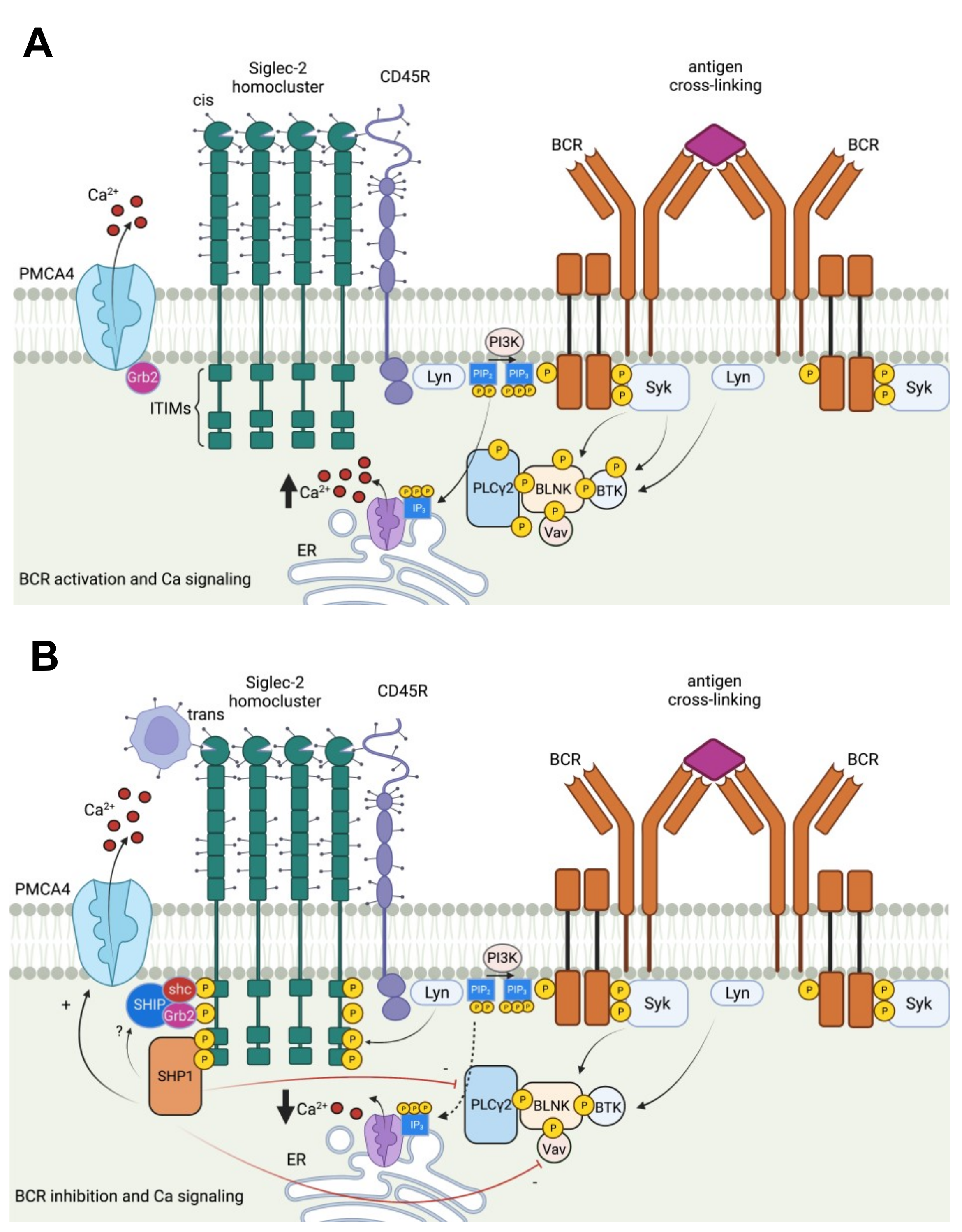
2.16. Siglec-Related Downstream Immune Signaling
3. Siglec-Directed Strategies to Counteract Tumor Immune Evasion
3.1. Anti-Siglec Based Immunotherapies
3.1.1. Naked Antibodies for Tumor Cell Lysis
3.1.2. Antibody Drug Conjugates (ADC) for Specific, Potent Tumor Targeting
3.1.3. Immunotoxins: Antibody-Derived Protein Toxin Conjugates
3.1.4. Bispecific Antibodies Targeting T Cells
3.1.5. Bi- or Tri-Specific Antibodies Targeting NK Cells
3.1.6. CD19/CD22-Directed CAR T-Cell Therapy
3.1.7. CD33-Directed CAR T-Cell Therapy
3.2. Sialic-Acid Ligands—Triumph over Siglec Immunosuppression
3.2.1. Designing High Affinity Sialic-Acid Ligands
3.2.2. Structural-Guided Sialoside Design
3.2.3. Sialoadhesin/CD22-Directed Liposomes in Cancer Therapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prendergast, G.C. Immune escape as a fundamental trait of cancer: Focus on IDO. Oncogene 2008, 27, 3889–3900. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nat. Cell Biol. 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Van Rooijen, N.; Weissman, I.L. CD47 Is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nat. Cell Biol. 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Nomura, T.; Huang, W.-C.; Zhau, H.; Josson, S.; Mimata, H.; Chung, L. β2-Microglobulin-mediated signaling as a target for cancer therapy. Anti Cancer Agents Med. Chem. 2014, 14, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L. Classification of advanced human cancers based on tumor immunity in the microenvironment (TIME) for cancer immunotherapy. JAMA Oncol. 2016, 2, 1403–1404. [Google Scholar] [CrossRef] [PubMed]
- Radvanyi, L.; Pilon-Thomas, S.; Peng, W.; Sarnaik, A.; Mulé, J.J.; Weber, J.; Hwu, P. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer—letter. Clin. Cancer Res. 2013, 19, 5541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büll, C.; Heise, T.; Adema, G.J.; Boltje, T.J. Sialic acid mimetics to target the sialic acid–Siglec axis. Trends Biochem. Sci. 2016, 41, 519–531. [Google Scholar] [CrossRef]
- Barrow, A.D.; Trowsdale, J. You say ITAM and I say ITIM, let’s call the whole thing off: The ambiguity of immunoreceptor signalling. Eur. J. Immunol. 2006, 36, 1646–1653. [Google Scholar] [CrossRef]
- Siddiqui, S.S.; Matar, R.; Merheb, M.; Hodeify, R.; Vazhappilly, C.G.; Marton, J.; Shamsuddin, S.A.; Al Zouabi, H. Siglecs in brain function and neurological disorders. Cells 2019, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Daly, J.; Carlsten, M.; O’Dwyer, M. Sugar free: Novel immunotherapeutic approaches targeting siglecs and sialic acids to enhance natural killer cell cytotoxicity against cancer. Front. Immunol. 2019, 10, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanczak, M.A.; Siddiqui, S.S.; Trefny, M.P.; Thommen, D.S.; Boligan, K.F.; Von Gunten, S.; Tzankov, A.; Tietze, L.; Lardinois, D.; Heinzelmann-Schwarz, V.; et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J. Clin. Investig. 2018, 128, 4912–4923. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, J.A.; Chang, A.T.; Youngblood, B.A.; Bochner, B.S. Eosinophil and mast cell Siglecs: From biology to drug target. J. Leukoc. Biol. 2020, 108, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Bärenwaldt, A.; Läubli, H. The sialoglycan-Siglec glyco-immune checkpoint—A target for improving innate and adaptive anti-cancer immunity. Expert Opin. Ther. Targets 2019, 23, 839–853. [Google Scholar] [CrossRef]
- Crocker, P.R.; Varki, A. Siglecs, sialic acids and innate immunity. Trends Immunol. 2001, 22, 337–342. [Google Scholar] [CrossRef]
- Fraschilla, I.; Pillai, S. Viewing Siglecs through the lens of tumor immunology. Immunol. Rev. 2017, 276, 178–191. [Google Scholar] [CrossRef] [Green Version]
- Läubli, H.; Varki, A. Sialic acid–binding immunoglobulin-like lectins (Siglecs) detect self-associated molecular patterns to regulate immune responses. Cell. Mol. Life Sci. 2020, 77, 593–605. [Google Scholar] [CrossRef]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. Siglec receptors and hiding plaques in Alzheimer’s disease. J. Mol. Med. 2009, 87, 697–701. [Google Scholar] [CrossRef]
- Lenza, M.P.; Atxabal, U.; Oyenarte, I.; Jiménez-Barbero, J.; Ereño-Orbea, J. Current Status on therapeutic molecules targeting Siglec receptors. Cells 2020, 9, 2691. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nat. Cell Biol. 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Crocker, P.; Kelm, S.; Dubois, C.; Martin, B.; McWilliam, A.; Shotton, D.; Paulson, J.; Gordon, S. Purification and properties of sialoadhesin, a sialic acid-binding receptor of murine tissue macrophages. EMBO J. 1991, 10, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much more than M1 and M2 macrophages, there are also CD169+ and TCR+ macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Takeya, M. Possible functions of CD169-positive sinus macrophages in lymph nodes in anti-tumor immune responses. Cancer Sci. 2017, 108, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, J.-H.; Kzhyshkowska, J.; Falkowski-Hansen, M.; Schledzewski, K.; Gratchev, A.; Mansmann, U.; Schmuttermaier, C.; Dippel, E.; Koenen, W.; Riedel, F.; et al. Differential expression of a gene signature for scavenger/lectin receptors by endothelial cells and macrophages in human lymph node sinuses, the primary sites of regional metastasis. J. Pathol. 2006, 208, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Van Dinther, D.; Veninga, H.; Iborra, S.; Borg, E.G.; Hoogterp, L.; Olesek, K.; Beijer, M.R.; Schetters, S.T.; Kalay, H.; Garcia-Vallejo, J.J.; et al. Functional CD169 on macrophages mediates interaction with dendritic cells for CD8+ T cell cross-priming. Cell Rep. 2018, 22, 1484–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, T.; Ohnishi, K.; Shiota, T.; Motoshima, T.; Sugiyama, Y.; Yatsuda, J.; Kamba, T.; Ishizaka, K.; Komohara, Y. CD 169-positive sinus macrophages in the lymph nodes determine bladder cancer prognosis. Cancer Sci. 2018, 109, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Kumamoto, K.; Tasaki, T.; Ohnishi, K.; Shibata, M.; Shimajiri, S.; Harada, M.; Komohara, Y.; Nakayama, T. CD169 Expression on lymph node macrophages predicts in patients with gastric cancer. Front. Oncol. 2021, 11, 636751. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Ohnishi, K.; Miyashita, A.; Nakahara, S.; Fujiwara, Y.; Horlad, H.; Motoshima, T.; Fukushima, S.; Jinnin, M.; Ihn, H.; et al. Prognostic significance of CD169+ lymph node sinus macrophages in patients with malignant melanoma. Cancer Immunol. Res. 2015, 3, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Choi, B.K. Siglec1-expressing subcapsular sinus macrophages provide soil for melanoma lymph node metastasis. eLife 2019, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.-Q.; Jiang, Z.-Z.; Li, L.; Wu, Y.; Zheng, L. CD169 identifies an anti-tumour macrophage subpopulation in human hepatocellular carcinoma. J. Pathol. 2016, 239, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Guo, X.; Wang, G.; Bi, Y.; Han, L.; Zhu, Q.; Qiu, C.; Tanaka, M.; Zhao, Y. Breast cancer cells promote CD169+ macrophage-associated immunosuppression through JAK2-mediated PD-L1 upregulation on macrophages. Int. Immunopharmacol. 2020, 78, 106012. [Google Scholar] [CrossRef]
- Jellusova, J.; Nitschke, L. Regulation of B cell functions by the sialic acid-binding receptors Siglec-G and CD22. Front. Immunol. 2012, 2, 96. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.J.; Linder, A.T.; Brandl, C.; Nitschke, L. B Cell Siglecs–news on signaling and its interplay with ligand binding. Front. Immunol. 2018, 9, 2820. [Google Scholar] [CrossRef] [PubMed]
- Poe, J.C.; Fujimoto, M.; Jansen, P.J.; Miller, A.S.; Tedder, T.F. CD22 forms a quaternary complex with SHIP, Grb2, and Shc: A pathway for regulation of B lymphocyte antigen receptor-induced calcium flux. J. Biol. Chem. 2000, 275, 17420–17427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Varki, A. Cell surface sialic acids do not affect primary CD22 interactions with CD45 and surface IgM nor the rate of constitutive CD22 endocytosis. Glycobiology 2004, 14, 939–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; Fitzgerald, D.J.; Barrett, D.M.; et al. Anti-CD22–chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Zhang, Y.; Zhao, H.; Wang, Y.; Liu, Y.; Liang, B.; Wang, X.; Xu, H.; Cui, J.; Wu, W.; et al. CD19/CD22 Dual-targeted CAR T-cell therapy for relapsed/refractory aggressive B-cell lymphoma: A safety and efficacy study. Cancer Immunol. Res. 2021, 9, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F.; Maffini, E.; Rondoni, M.; Massari, E.; Faini, A.C.; Malavasi, F. CD22 expression in B-cell acute lymphoblastic leukemia: Biological significance and implications for inotuzumab therapy in adults. Cancers 2020, 12, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, L.M.; Barman, S.; Shao, C.; Poe, J.C.; Venturi, G.M.; Shelton, J.M.; Pop, I.V.; Gerber, D.; Girard, L.; Liu, X.-Y.; et al. A reevaluation of CD22 expression in human lung cancer. Cancer Res. 2014, 74, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masih, K.E.; Ligon, J.A.; Yates, B.; Shalabi, H.; Little, L.; Islam, Z.; Ombrello, A.K.; Inglefield, J.; Nussenblatt, V.; Manion, M.; et al. Consequences of hemophagocytic lymphohistiocytosis-like cytokine release syndrome toxicities and concurrent bacteremia. Pediatr. Blood Cancer 2021, 68, e29247. [Google Scholar] [CrossRef]
- Lichtenstein, D.A.; Schischlik, F.; Shao, L.; Steinberg, S.M.; Yates, B.; Wang, H.-W.; Wang, Y.; Inglefield, J.; Florea, A.D.; Ceppi, F.; et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T-cells. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef]
- Siddiqui, S.; Springer, S.A.; Verhagen, A.; Sundaramurthy, V.; Alisson-Silva, F.; Jiang, W.; Ghosh, P.; Varki, A. The Alzheimer’s disease–protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J. Biol. Chem. 2017, 292, 15312–15320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 Expression and gentuzumab ozogamicin in acute myeloid leukemia: Two sides of the same coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Rudd, C.E. Tyrosine phosphatase SHP-2 binding to CTLA-4: Absence of direct YVKM/YFIP motif recognition. Biochem. Biophys. Res. Commun. 2000, 269, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.; Riley, J.L. SHP-1 and SHP-2 Associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, V.H.J.; Marvelde, J.G.T.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; Van Dongen, J.J.M. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B. The role of CD33 as therapeutic target in acute myeloid leukemia. Expert Opin. Ther. Targets 2014, 18, 715–718. [Google Scholar] [CrossRef]
- Stokke, J.L.; Bhojwani, D. Antibody–drug conjugates for the treatment of acute pediatric leukemia. J. Clin. Med. 2021, 10, 3556. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Yu, S.-F.; Del Rosario, G.; Leong, S.R.; Lee, G.Y.; Vij, R.; Chiu, C.P.; Liang, W.-C.; Wu, Y.; Chalouni, C.; et al. An anti–CLL-1 antibody–drug conjugate for the treatment of acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Allen, K.J.H.; Dawicki, W.; Geoghegan, E.M.; Ludwig, D.L.; Dadachova, E. 225Ac-labeled CD33-targeting antibody reverses resistance to Bcl-2 inhibitor venetoclax in acute myeloid leukemia models. Cancer Med. 2021, 10, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Frail, D.F.; Braun, P.E. Two developmentally regulated messenger RNAs differing in their coding region may exist for the myelin-associated glycoprotein. J. Biol. Chem. 1984, 259, 14857–14862. [Google Scholar] [CrossRef]
- Lai, C.; Brow, M.A.; Nave, K.A.; Noronha, A.B.; Quarles, R.H.; Bloom, F.E.; Milner, R.J.; Sutcliffe, J.G. Two forms of 1B236/myelin-associated glycoprotein, a cell adhesion molecule for postnatal neural development, are produced by alternative splicing. Proc. Natl. Acad. Sci. USA 1987, 84, 4337–4341. [Google Scholar] [CrossRef] [Green Version]
- Jaccard, A. Monoclonal IgM and neuropathy: Not always anti-MAG. Blood 2020, 136, 2366–2367. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Hirano, M.; Kuwahara, M.; Samukawa, M.; Takada, K.; Morise, J.; Yabuno, K.; Oka, S.; Kusunoki, S. Binding specificity of anti-HNK-1 IgM M-protein in anti-MAG neuropathy: Possible clinical relevance. Neurosci. Res. 2015, 91, 63–68. [Google Scholar] [CrossRef]
- Swanson, B.J.; McDermott, K.M.; Singh, P.K.; Eggers, J.P.; Crocker, P.; Hollingsworth, M.A. MUC1 Is a counter-receptor for myelin-associated glycoprotein (Siglec-4a) and their interaction contributes to adhesion in pancreatic cancer perineural invasion. Cancer Res. 2007, 67, 10222–10229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, D.; Henze, J.; Pfeifer, R.; Schleicher, A.; Brauner, J.; Mockel-Tenbrinck, N.; Barth, C.; Gudert, D.; Al Rawashdeh, W.; Johnston, I.C.D.; et al. A novel Siglec-4 derived spacer improves the functionality of CAR T cells against membrane-proximal epitopes. Front. Immunol. 2020, 11, 1704. [Google Scholar] [CrossRef] [PubMed]
- Brinkman-Van Der Linden, E.C.M.; Varki, A. New aspects of siglec binding specificities, including the significance of fucosylation and of the Sialyl-Tn epitope. J. Biol. Chem. 2000, 275, 8625–8632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.R.; Fong, J.J.; Carlin, A.F.; Busch, T.D.; Linden, R.; Angata, T.; Areschoug, T.; Parast, M.; Varki, N.; Murray, J.; et al. Siglec-5 and Siglec-14 are polymorphic paired receptors that modulate neutrophil and amnion signaling responses to group B Streptococcus. J. Exp. Med. 2014, 211, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Freeman, S.; Attrill, H.; Clarke, R.G.; Crocker, P. Siglec-5 (CD170) can mediate inhibitory signaling in the absence of immunoreceptor tyrosine-based inhibitory motif phosphorylation. J. Biol. Chem. 2005, 280, 19843–19851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepin, M.; Mezouar, S.; Pegon, J.; Muczynski, V.; Adam, F.; Bianchini, E.P.; Bazaa, A.; Proulle, V.; Rupin, A.; Paysant, J.; et al. Soluble Siglec-5 associates to PSGL-1 and displays anti-inflammatory activity. Sci. Rep. 2016, 6, 37953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgo, P.; Denning-Kendall, P.A.; Erickson-Miller, C.L.; Singha, S.; Evely, R.; Hows, J.M.; Freeman, S.D. Identification of the CD33-related Siglec receptor, Siglec-5 (CD170), as a useful marker in both normal myelopoiesis and acute myeloid leukaemias. Br. J. Haematol. 2003, 123, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Montalbán-Hernández, K.; Cantero-Cid, R.; Lozano-Rodríguez, R.; Pascual-Iglesias, A.; Avendaño-Ortiz, J.; Casalvilla-Dueñas, J.; Pérez, G.B.; Guevara, J.; Marcano, C.; Barragán, C.; et al. Soluble SIGLEC5: A new prognosis marker in colorectal cancer patients. Cancers 2021, 13, 3896. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Hurtado-Ziola, N.; Gagneux, P.; Varki, A. Loss of Siglec expression on T lymphocytes during human evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 7765–7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuchkovska, A.; Iwashima, M. Siglec 5—A novel checkpoint receptor in T cells. J. Immunol. 2020, 204, 11–78. [Google Scholar]
- Yamanaka, M.; Kato, Y.; Angata, T.; Narimatsu, H. Deletion polymorphism of SIGLEC14 and its functional implications. Glycobiology 2009, 19, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-M.; Riestra, A.; Ali, S.R.; Fong, J.J.; Liu, J.Z.; Hughes, G.; Varki, A.; Nizet, V. Siglec-14 enhances NLRP3-inflammasome activation in macrophages. J. Innate Immun. 2020, 12, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-C.J.; Low, P.-Y.; Wang, I.; Hsu, S.-T.D.; Angata, T. Soluble Siglec-14 glycan-recognition protein is generated by alternative splicing and suppresses myeloid inflammatory responses. J. Biol. Chem. 2018, 293, 19645–19658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; der Linden, E.C.M.B.-V.; Altmann, S.W.; Gish, K.; Balasubramanian, S.; Timans, J.C.; Peterson, D.; Bell, M.P.; Bazan, J.F.; Varki, A.; et al. OB-BP1/Siglec-6. J. Biol. Chem. 1999, 274, 22729–22738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, H.; Myers, A.; Matsumoto, K.; Crocker, P.R.; Saito, H.; Bochner, B.S. Alteration and acquisition of Siglecs during in vitro maturation of CD34+ progenitors into human mast cells. Allergy 2006, 61, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Issa, F. Single-cell RNA-Seq reveals new types of human blood dendritic cells, monocytes, and progenitor. Transplantation 2017, 101, 1955–1956. [Google Scholar] [CrossRef]
- Blixt, O.; Collins, B.E.; Nieuwenhof, I.M.V.D.; Crocker, P.; Paulson, J.C. Sialoside specificity of the siglec family assessed using novel multivalent probes: Identification of potent inhibitors of myelin-associated glycoprotein. J. Biol. Chem. 2003, 278, 31007–31019. [Google Scholar] [CrossRef] [Green Version]
- Benmerzoug, S.; Chevalier, M.F.; Verardo, M.; Nguyen, S.; Cesson, V.; Schneider, A.K.; Dartiguenave, F.; Rodrigues-Dias, S.-C.; Lucca, I.; Jichlinski, P.; et al. Siglec-6 as a new potential immune checkpoint for bladder cancer patients. Eur. Urol. Focus 2021, 2021, 10–13. [Google Scholar] [CrossRef]
- Kovalovsky, D.; Yoon, J.H.; Cyr, M.G.; Simon, S.; Voynova, E.; Rader, C.; Wiestner, A.; Alejo, J.; Pittaluga, S.; Gress, R.E. Siglec-6 is a target for chimeric antigen receptor T-cell treatment of chronic lymphocytic leukemia. Leukemia 2021, 35, 2581–2591. [Google Scholar] [CrossRef]
- Rosenstock, P.; Horstkorte, R.; Gnanapragassam, V.S.; Harth, J.; Kielstein, H. Siglec-7 expression is reduced on a natural killer (NK) cell subset of obese humans. Immunol. Res. 2017, 65, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoll, G.; Ni, J.; Liu, D.; Klenerman, P.; Munday, J.; Dubock, S.; Mattei, M.-G.; Crocker, P.R. Identification and characterization of a novel Siglec, Siglec-7, expressed by human natural killer cells and monocytes. J. Biol. Chem. 1999, 274, 34089–34095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikehara, Y.; Ikehara, S.; Paulson, J.C. Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J. Biol. Chem. 2004, 279, 43117–43125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaji, T.; Teranishi, T.; Alphey, M.S.; Crocker, P.; Hashimoto, Y. A small region of the natural killer cell receptor, Siglec-7, is responsible for its preferred binding to α2,8-disialyl and branched α2,6-sialyl residues: A comparison with Siglec-9. J. Biol. Chem. 2002, 277, 6324–6332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, N.; Yasuda, Y.; Yoshimura, A.; Goshima, A.; Crocker, P.R.; Vergoten, G.; Nishiura, Y.; Takahashi, T.; Hanashima, S.; Matsumoto, K.; et al. Discovery of a new sialic acid binding region that regulates Siglec-7. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Hong, S.; Yu, C.; Rodrigues, E.; Shi, Y.; Chen, H.; Wang, P.; Chapla, D.G.; Gao, T.; Zhuang, R.; Moremen, K.W.; et al. Modulation of Siglec-7 signaling via in situ-created high-affinity cis-ligands. ACS Central Sci. 2021, 7, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, G.; Avril, T.; Lock, K.; Furukawa, K.; Bovin, N.; Crocker, P.R. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur. J. Immunol. 2003, 33, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Ravetch, J.V.; Lanier, L.L. Immune inhibitory receptors. Science 2000, 290, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wang, S.; Yang, L.; Jiang, L.; Li, J.; Wang, X. Reduced Siglec-7 expression on NK cells predicts NK cell dysfunction in primary hepatocellular carcinoma. Clin. Exp. Immunol. 2020, 201, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Varchetta, S.; Brunetta, E.; Roberto, A.; Mikulak, J.; Hudspeth, K.L.; Mondelli, M.; Mavilio, D. Engagement of Siglec-7 receptor induces a pro-inflammatory response selectively in monocytes. PLoS ONE 2012, 7, e45821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, E.; Boelaars, K.; Brown, K.; Li, R.J.E.; Kruijssen, L.; Bruijns, S.C.M.; van Ee, T.; Schetters, S.T.T.; Crommentuijn, M.H.W.; van der Horst, J.C.; et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Yamada, K.; Hazama, S.; Suzuki, N.; Xu, M.; Nakagami, Y.; Fujiwara, N.; Tsunedomi, R.; Yoshida, S.; Tomochika, S.; Matsukuma, S.; et al. Siglec-7 is a predictive biomarker for the efficacy of cancer vaccination against metastatic colorectal cancer. Oncol. Lett. 2020, 21, 1. [Google Scholar] [CrossRef]
- Meril, S.; Harush, O.; Reboh, Y.; Matikhina, T.; Barliya, T.; Cohen, C.J. Targeting glycosylated antigens on cancer cells using siglec-7/9-based CAR T-cells. Mol. Carcinog. 2020, 59, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Plitt, J.; Bochner, B.S. Human eosinophils express two Siglec-8 splice variants. J. Allergy Clin. Immunol. 2002, 109, 176. [Google Scholar] [CrossRef]
- Rapoport, E.M.; Pazynina, G.V.; Sablina, M.A.; Crocker, P.R.; Bovin, N.V. Probing sialic acid binding Ig-like lectins (siglecs) with sulfated oligosaccharides. Biochemistry 2006, 71, 496–504. [Google Scholar] [CrossRef]
- Morris, S.; Ahmad, N.; André, S.; Kaltner, H.; Gabius, H.-J.; Brenowitz, M.; Brewer, F. Quaternary solution structures of galectins-1, -3, and -7. Glycobiology 2003, 14, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Pröpster, J.M.; Yang, F.; Rabbani, S.; Ernst, B.; Allain, F.H.-T.; Schubert, M. Structural basis for sulfation-dependent self-glycan recognition by the human immune-inhibitory receptor Siglec-8. Proc. Natl. Acad. Sci. USA 2016, 113, E4170–E4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellon, E.S.; Peterson, K.A.; Murray, J.A.; Falk, G.W.; Gonsalves, N.; Chehade, M.; Genta, R.M.; Leung, J.; Khoury, P.; Klion, A.D.; et al. Anti–Siglec-8 antibody for eosinophilic gastritis and duodenitis. N. Engl. J. Med. 2020, 383, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.A.; Leung, J.; Falahati, R.; Williams, J.; Schanin, J.; Brock, E.C.; Singh, B.; Chang, A.T.; O’Sullivan, J.A.; Schleimer, R.P.; et al. Discovery, function, and therapeutic targeting of Siglec-8. Cells 2020, 10, 19. [Google Scholar] [CrossRef]
- Ou, C.; Liu, L.; Wang, J.; Dai, S.; Qu, Y.; Xiong, Y.; Xi, W.; Xu, J.; Guo, J. Enhancement of Siglec-8 expression predicts adverse prognosis in patients with clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 607.e1–607.e8. [Google Scholar] [CrossRef]
- Haas, Q.; Boligan, K.F.; Jandus, C.; Schneider, C.; Simillion, C.; Stanczak, M.; Haubitz, M.; Jafari, S.M.S.; Zippelius, A.; Baerlocher, G.M.; et al. Siglec-9 regulates an effector memory CD8+ T-cell subset that congregates in the melanoma tumor microenvironment. Cancer Immunol. Res. 2019, 7, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Bai, F.-F.; Han, L.; Zhu, J.; Zheng, T.; Zhu, Z.; Zhou, L.-F. Targeting neutrophils in severe asthma via Siglec-9. Int. Arch. Allergy Immunol. 2018, 175, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ma, X.; Su, D.; Zhang, Y.; Yu, L.; Jiang, F.; Zhou, X.; Feng, Y.; Ma, F. The roles of Siglec7 and Siglec9 on natural killer cells in virus infection and tumour progression. J. Immunol. Res. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Yu, H.; Gonzalez-Gil, A.; Wei, Y.; Fernandes, S.M.; Porell, R.N.; Vajn, K.; Paulson, J.C.; Nycholat, C.M.; Schnaar, R.L. Siglec-8 and Siglec-9 binding specificities and endogenous airway ligand distributions and properties. Glycobiology 2017, 27, 657–668. [Google Scholar] [CrossRef]
- Von Gunten, S.; Yousefi, S.; Seitz, M.; Jakob, S.M.; Schaffner, T.; Seger, R.; Takala, J.; Villiger, P.M.; Simon, H.-U. Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the proinflammatory cytokine environment. Blood 2005, 106, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Läubli, H.; Pearce, O.; Schwarz, F.; Siddiqui, S.; Deng, L.; Stanczak, M.; Deng, L.; Verhagen, A.; Secrest, P.; Lusk, C.; et al. Engagement of myelomonocytic Siglecs by tumor-associated ligands modulates the innate immune response to cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14211–14216. [Google Scholar] [CrossRef] [Green Version]
- Jandus, C.; Boligan, K.F.; Chijioke, O.; Liu, H.; Dahlhaus, M.; Démoulins, T.; Schneider, C.; Wehrli, M.; Hunger, R.E.; Baerlocher, G.M.; et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell–dependent tumor immunosurveillance. J. Clin. Investig. 2014, 124, 1810–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatson, R.; Graham, R.; Freile, F.G.; Cozzetto, D.; Kannambath, S.; Pfeifer, E.; Woodman, N.; Owen, J.; Nuamah, R.; Mandel, U.; et al. Cancer-associated hypersialylated MUC1 drives the differentiation of human monocytes into macrophages with a pathogenic phenotype. Commun. Biol. 2020, 3, 1–15. [Google Scholar] [CrossRef]
- Burchell, J.; Poulsom, R.; Hanby, A.; Whitehouse, C.; Cooper, L.; Clausen, H.; Miles, D.; Taylor-Papadimitriou, J. An 2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology 1999, 9, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Hoballah, J.; Fritz-Klaus, R.; Al-Johani, L.; Brooker, J.; Patankar, M.S. Characterization of cell-bound CA125 on immune cell subtypes. Cancers 2021, 13, 2072. [Google Scholar]
- Tomioka, Y.; Morimatsu, M.; Nishijima, K.-I.; Usui, T.; Yamamoto, S.; Suyama, H.; Ozaki, K.; Ito, T.; Ono, E. A soluble form of Siglec-9 provides an antitumor benefit against mammary tumor cells expressing MUC1 in transgenic mice. Biochem. Biophys. Res. Commun. 2014, 450, 532–537. [Google Scholar] [CrossRef]
- Ibarlucea-Benitez, I.; Weitzenfeld, P.; Smith, P.; Ravetch, J.V. Siglecs-7/9 function as inhibitory immune checkpoints in vivo and can be targeted to enhance therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2021, 118, e2107424118. [Google Scholar] [CrossRef]
- Biedermann, B.; Gil, D.; Bowen, D.T.; Crocker, P.R. Analysis of the CD33-related siglec family reveals that Siglec-9 is an endocytic receptor expressed on subsets of acute myeloid leukemia cells and absent from normal hematopoietic progenitors. Leukemia Res. 2007, 31, 211–220. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, X.; Tao, K.; Shi, L.; Li, W.; Wang, G.; Wu, K. Siglec-10 is associated with survival and natural killer cell dysfunction in hepatocellular carcinoma. J. Surg. Res. 2015, 194, 107–113. [Google Scholar] [CrossRef]
- Bandala-Sanchez, E.; Bediaga, N.G.; Goddard-Borger, E.D.; Ngui, K.; Naselli, G.; Stone, N.L.; Neale, A.M.; Pearce, L.A.; Wardak, A.; Czabotar, P.; et al. CD52 glycan binds the proinflammatory B box of HMGB1 to engage the Siglec-10 receptor and suppress human T cell function. Proc. Natl. Acad. Sci. USA 2019, 116, 7592–7593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandala-Sanchez, E.; Bediaga, N.; Naselli, G.; Neale, A.; Harrison, L. Siglec-10 expression is up-regulated in activated human CD4+ T cells. Hum. Immunol. 2020, 81, 101–104. [Google Scholar] [CrossRef]
- Bandala-Sanchez, E.; Zhang, Y.; Reinwald, S.; Dromey, J.A.; Lee, B.-H.; Qian, J.; Böhmer, R.M.; Harrison, L.C. T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nat. Immunol. 2013, 14, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Dente, L.; Riither, U.; Tripodi, M.; Wagner, E.F.; Cortese, R. Expression of human alpha1-acid glycoproteln genes in cultured cells and in transgenic mice. Genes Dev. 1988, 2, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Board, P.G.; Matsushita, K.; Tanaka, H.; Matsuyama, T.; Matsuda, T. α1Acid glycoprotein expression in human leukocytes: Possible correlation between α1-acid glycoprotein and inflammatory cytokines in rheumatoid arthritis. Inflammation 1993, 17, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, G.-Y.; Zheng, P. CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol. 2009, 30, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.-Y.; Tang, J.; Zheng, P.; Liu, Y. CD24 and Siglec-10 selectively repress tissue damage–induced immune responses. Science 2009, 323, 1722–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.-Y.; Chen, X.; King, S.; Cavassani, K.A.; Cheng, J.; Zheng, X.; Cao, H.; Yu, H.; Qu, J.; Fang, D.; et al. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat. Biotechnol. 2011, 29, 428–435. [Google Scholar] [CrossRef]
- Xiao, N.; Zhu, X.; Li, K.; Chen, Y.; Liu, X.; Xu, B.; Lei, M.; Xu, J.; Sun, H.-C. Blocking siglec-10hi tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp. Hematol. Oncol. 2021, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, J.; Zhuo, Q.; Zhang, J.; Xie, J.; Han, S.; Zhao, S. Malignant ascite-derived extracellular vesicles inhibit T cell activity by upregulating Siglec-10 expression. Cancer Manag. Res. 2019, 11, 7123–7134. [Google Scholar] [CrossRef] [Green Version]
- Rashidi, M.; Bandala-Sanchez, E.; Lawlor, K.; Zhang, Y.; Neale, A.M.; Vijayaraj, S.L.; O’Donoghue, R.; Wentworth, J.M.; Adams, T.; Vince, J.E.; et al. CD52 inhibits toll-like receptor activation of NF-κB and triggers apoptosis to suppress inflammation. Cell Death Differ. 2018, 25, 392–405. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Nakahata, S.; Yamakawa, N.; Kaneda-Nakashima, K.; Ichihara, E.; Suekane, A.; Morishita, K. CD52 as a molecular target for immunotherapy to treat acute myeloid leukemia with high EVI1 expression. Leukemia 2011, 25, 921–931. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, G.; Sui, Y.; Yang, Z.; Chu, Y.; Tang, H.; Guo, B.; Zhang, C.; Wu, C. CD52 Is a prognostic biomarker and associated with tumor microenvironment in breast cancer. Front. Genet. 2020, 11, 1350. [Google Scholar] [CrossRef]
- Wang, X.; Chow, R.; Deng, L.; Anderson, D.; Weidner, N.; Godwin, A.K.; Bewtra, C.; Zlotnik, A.; Bui, J.; Varki, A.; et al. Expression of Siglec-11 by human and chimpanzee ovarian stromal cells, with uniquely human ligands: Implications for human ovarian physiology and pathology. Glycobiology 2011, 21, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, T.; Khedri, Z.; Schwarz, F.; Landig, C.; Liang, S.-Y.; Toshiyuki, H.; Chen, X.; Fujito, N.T.; Satta, Y.; Varki, A.; et al. Coevolution of Siglec-11 and Siglec-16 via gene conversion in primates. BMC Evol. Biol. 2017, 17, 228. [Google Scholar] [CrossRef]
- Wang, X.; Mitra, N.; Cruz, P.; Deng, L.; Varki, N.; Angata, T.; Green, E.D.; Mullikin, J.; Hayakawa, T.; Varki, A.; et al. Evolution of Siglec-11 and Siglec-16 genes in hominins. Mol. Biol. Evol. 2012, 29, 2073–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Lakner, U.; de Bono, B.; Traherne, J.A.; Trowsdale, J.; Barrow, A.D. SIGLEC16 encodes a DAP12-associated receptor expressed in macrophages that evolved from its inhibitory counterpart SIGLEC11 and has functional and non-functional alleles in humans. Eur. J. Immunol. 2008, 38, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Hane, M.; Chen, D.Y.; Varki, A. Human-specific microglial Siglec-11 transcript variant has the potential to affect polysialic acid-mediated brain functions at a distance. Glycobiology 2021, 31, 231–242. [Google Scholar] [CrossRef]
- Wang, Y.; Neumann, H. Alleviation of Neurotoxicity by microglial human Siglec-11. J. Neurosci. 2010, 30, 3482–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnartz-Gerlach, B.; Kopatz, J.; Neumann, H. Siglec functions of microglia. Glycobiology 2014, 24, 794–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wielgat, P.; Wawrusiewicz-Kurylonek, N.; Czarnomysy, R.; Rogowski, K.; Bielawski, K.; Car, H. The paired siglecs in brain tumours therapy: The immunomodulatory effect of dexamethasone and temozolomide in human glioma in vitro model. Int. J. Mol. Sci. 2021, 22, 1791. [Google Scholar] [CrossRef]
- Põlajeva, J.; Sjösten, A.M.; Lager, N.; Kastemar, M.; Waern, I.; Alafuzoff, I.; Smits, A.; Westermark, B.; Pejler, G.; Uhrbom, L.; et al. Mast cell accumulation in glioblastoma with a potential role for stem cell factor and chemokine CXCL12. PLoS ONE 2011, 6, e25222. [Google Scholar] [CrossRef] [Green Version]
- Mitra, N.; Banda, K.; Altheide, T.K.; Schaffer, L.; Johnson-Pais, T.L.; Beuten, J.; Leach, R.; Angata, T.; Varki, N.; Varki, A. SIGLEC12, a human-specific segregating (pseudo)gene, encodes a signaling molecule expressed in prostate carcinomas. J. Biol. Chem. 2011, 286, 23003–23011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, S.S.; Vaill, M.; Do, R.; Khan, N.; Verhagen, A.L.; Zhang, W.; Lenz, H.; Johnson-Pais, T.L.; Leach, R.J.; Fraser, G.; et al. Human-specific polymorphic pseudogenization of SIGLEC12 protects against advanced cancer progression. FASEB BioAdvances 2021, 3, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mitra, N.; Secundino, I.; Banda, K.; Cruz, P.; Padler-Karavani, V.; Verhagen, A.; Reid, C.; Lari, M.; Rizzi, E.; et al. Specific inactivation of two immunomodulatory SIGLEC genes during human evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 9935–9940. [Google Scholar] [CrossRef] [Green Version]
- Angata, T.; Tabuchi, Y.; Nakamura, K.; Nakamura, M. Siglec-15: An immune system Siglec conserved throughout vertebrate evolution. Glycobiology 2007, 17, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019, 25, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. DAP10- and DAP12-associated receptors in innate immunity. Immunol. Rev. 2008, 227, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantuano, N.R.; Natoli, M.; Zippelius, A.; Läubli, H. Tumor-associated carbohydrates and immunomodulatory lectins as targets for cancer immunotherapy. J. Immunother. Cancer 2020, 8, e001222. [Google Scholar] [CrossRef]
- Julien, S.; Videira, P.A.; Delannoy, P. Sialyl-Tn in cancer: (How) did we miss the target? Biomolecules 2012, 2, 435–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briard, J.G.; Jiang, H.; Moremen, K.W.; Macauley, M.S.; Wu, P. Cell-based glycan arrays for probing glycan–glycan binding protein interactions. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, B.; Wang, X.; Zeng, Z.; Huang, Z.; Zhang, L.; Wei, F.; Ren, X.; Yang, L. Expression signature, prognosis value, and immune characteristics of Siglec-15 identified by pan-cancer analysis. OncoImmunology 2020, 9, 1807291. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, R.; Ohtsubo, K.; Takamatsu, S.; Taniguchi, N.; Angata, T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF- secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology 2013, 23, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.; Stadanlick, J.; Annunziata, M.J.; Rao, A.S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Cantu, E.; Danet-Desnoyers, G.; Ra, H.-J.; et al. Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-T.; Huang, Z.-Z.; Chen, Y.-B.; Yao, H.-Y.; Ke, Z.-H.; He, X.-X.; Qiu, M.-J.; Wang, M.-M.; Xiong, Z.-F.; Yang, S.-L. Integrative analysis of Siglec-15 mRNA in human cancers based on data mining. J. Cancer 2020, 11, 2453–2464. [Google Scholar] [CrossRef]
- Murugesan, G.; Correia, V.G.; Palma, A.S.; Chai, W.; Li, C.; Feizi, T.; Martin, E.; Laux, B.; Franz, A.; Fuchs, K.; et al. Siglec-15 recognition of sialoglycans on tumor cell lines can occur independently of sialyl Tn antigen expression. Glycobiology 2020, 31, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.-K.; Zhang, G.-C.; Chen, W.; Qi, L.-L.; Xie, M.-F.; Zhang, Y.-Y.; Wang, L.; Zhang, Q. Siglec-15 promotes tumor progression in osteosarcoma via DUSP1/MAPK pathway. Front. Oncol. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ji, Z.; Wu, B.; Huang, S.; Chen, Q.; Chen, X.; Wei, Y.; Jiang, J. Siglec-15 promotes the migration of liver cancer cells by repressing lysosomal degradation of CD44. FEBS Lett. 2021, 595, 2290–2302. [Google Scholar] [CrossRef] [PubMed]
- Fudaba, H.; Momii, Y.; Hirakawa, T.; Onishi, K.; Asou, D.; Matsushita, W.; Kawasaki, Y.; Sugita, K.; Fujiki, M. Sialic acid-binding immunoglobulin-like lectin-15 expression on peritumoral macrophages is a favorable prognostic factor for primary central nervous system lymphoma patients. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, Q.; Sanmanmed, M.F.; Wang, J. Siglec-15 as an emerging target for next-generation cancer immunotherapy. Clin. Cancer Res. 2021, 27, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Veillette, A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat. Immunol. 2012, 13, 439–447. [Google Scholar] [CrossRef]
- Walter, R.B.; Raden, B.W.; Zeng, R.; Häusermann, P.; Bernstein, I.D.; Cooper, J.A. ITIM-dependent endocytosis of CD33-related Siglecs: Role of intracellular domain, tyrosine phosphorylation, and the tyrosine phosphatases, Shp1 and Shp2. J. Leukoc. Biol. 2008, 83, 200–211. [Google Scholar] [CrossRef]
- Dustin, L.B.; Plas, D.R.; Wong, J.; Hu, Y.T.; Soto, C.; Chan, A.C.; Thomas, M.L. Expression of dominant-negative src-homology domain 2-containing protein tyrosine phosphatase-1 results in increased Syk tyrosine kinase activity and B cell activation. J. Immunol. 1999, 162, 2717–2724. [Google Scholar] [PubMed]
- Au-Yeung, B.; Deindl, S.; Hsu, L.-Y.; Palacios, E.H.; Levin, S.E.; Kuriyan, J.; Weiss, A. The structure, regulation, and function of ZAP-70. Immunol. Rev. 2009, 228, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Chiang, G.G.; Sefton, B.M. Specific dephosphorylation of the lck tyrosine protein kinase at Tyr-394 by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 2001, 276, 23173–23178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, M.; Wahid, M.; Khan, F. Regulation of peripheral and central immunity: Understanding the role of Src homology 2 domain-containing tyrosine phosphatases, SHP-1 & SHP-2. Immunobiology 2020, 225, 151847. [Google Scholar] [CrossRef]
- Hudson, C.A.; Christophi, G.P.; Cao, L.; Gruber, R.C.; Massa, P.T. Regulation of avoidant behaviors and pain by the anti-inflammatory tyrosine phosphatase SHP-1. Neuron Glia Biol. 2006, 2, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Collins, C.A.; Wu, P.; Brown, E.J. Protein tyrosine phosphatase SHP-1 positively regulates TLR-induced IL-12p40 production in macrophages through inhibition of phosphatidylinositol 3-kinase. J. Leukoc. Biol. 2010, 87, 845–855. [Google Scholar] [CrossRef]
- Massa, P.T.; Wu, C. Increased inducible activation of NF-κB and responsive genes in astrocytes deficient in the protein tyrosine phosphatase SHP-1. J. Interf. Cytokine Res. 1998, 18, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, C.C.; Pao, L.I.; Blattman, J.N.; Greenberg, P.D. SHP-1 in T cells limits the production of CD8 effector cells without impacting the formation of long-lived central memory cells. J. Immunol. 2010, 185, 3256–3267. [Google Scholar] [CrossRef] [Green Version]
- Pao, L.I.; Lam, K.-P.; Henderson, J.M.; Kutok, J.L.; Alimzhanov, M.; Nitschke, L.; Thomas, M.L.; Neel, B.G.; Rajewsky, K. B cell-specific deletion of protein-tyrosine phosphatase Shp1 Promotes B-1a cell development and causes systemic autoimmunity. Immunity 2007, 27, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Matsumoto, K.; Nishida, E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor sprouty. J. Biol. Chem. 2004, 279, 22992–22995. [Google Scholar] [CrossRef] [Green Version]
- Bollu, L.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular pathways: Targeting protein tyrosine phosphatases in cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef] [Green Version]
- Tajan, M.; Serra, A.D.R.; Valet, P.; Edouard, T.; Yart, A. SHP2 sails from physiology to pathology. Eur. J. Med Genet. 2015, 58, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Wang, C.; Sholy, C.; Cao, M.; Kang, X. Strategy for leukemia treatment targeting SHP-1,2 and SHIP. Front. Cell Dev. Biol. 2021, 9, 730400. [Google Scholar] [CrossRef]
- Dong, S.; Li, F.-Q.; Zhang, Q.; Lv, K.-Z.; Yang, H.-L.; Gao, Y.; Yu, J.-R. Expression and clinical significance of SHP2 in gastric cancer. J. Int. Med Res. 2012, 40, 2083–2089. [Google Scholar] [CrossRef]
- Yuan, E.A.Y.; Fan, Y.; Gao, Z.; Sun, X.; Zhang, H.; Wang, Z.; Cui, Y.; Song, W.; Wang, Z.; Zhang, F.; et al. SHP2 promotes proliferation of breast cancer cells through regulating Cyclin D1 stability via the PI3K/AKT/GSK3β signaling pathway. Cancer Biol. Med. 2020, 17, 707–725. [Google Scholar] [CrossRef]
- Dong, L.; Han, D.; Meng, X.; Xu, M.; Zheng, C.; Xia, Q. Activating mutation of SHP2 establishes a tumorigenic phonotype through cell-autonomous and non-cell-autonomous mechanisms. Front. Cell Dev. Biol. 2021, 9, 630712. [Google Scholar] [CrossRef]
- Serra-Nedelec, A.D.R.; Edouard, T.; Treguer, K.; Tajan, M.; Araki, T.; Dance, M.; Mus, M.; Montagner, A.; Tauber, M.; Salles, J.-P.; et al. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc. Natl. Acad. Sci. USA 2012, 109, 4257–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, S.J.; Morgan, N.M.; Elliott, J.; Burrows, J.F.; Scott, C.J.; McVicar, D.W.; Johnston, J.A. CD33 responses are blocked by SOCS3 through accelerated proteasomal-mediated turnover. Blood 2006, 109, 1061–1068. [Google Scholar] [CrossRef]
- Dayer, J.-M.; Chizzolini, C. Constitutive repressor activity of CD33 on human monocytes requires sialic acid recognition and phosphoinositide 3-kinase-mediated intracellular signaling. Eur. J. Immunol. 2004, 35, 243–251. [Google Scholar] [CrossRef]
- Angata, T. Siglecs that associate with DAP12. Adv. Exp. Med. Biol. 2020, 1204, 215–230. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niogret, C.; Birchmeier, W.; Guarda, G. SHP-2 in lymphocytes’ cytokine and inhibitory receptor signaling. Front. Immunol. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Dempke, W.C.; Uciechowski, P.; Fenchel, K.; Chevassut, T. Targeting SHP-1, 2 and SHIP pathways: A novel strategy for cancer treatment? Oncology 2018, 95, 257–269. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.-Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [Green Version]
- Li, X.J.; Goodwin, C.B.; Nabinger, S.C.; Richine, B.M.; Yang, Z.; Hanenberg, H.; Ohnishi, H.; Matozaki, T.; Feng, G.-S.; Chan, R.J. Protein-tyrosine phosphatase Shp2 positively regulates macrophage oxidative burst. J. Biol. Chem. 2015, 290, 3894–3909. [Google Scholar] [CrossRef] [Green Version]
- Heun, Y.; Pircher, J.; Czermak, T.; Bluem, P.; Hupel, G.; Bohmer, M.; Kraemer, B.F.; Pogoda, K.; Pfeifer, A.; Woernle, M.; et al. Inactivation of the tyrosine phosphatase SHP-2 drives vascular dysfunction in Sepsis. EBioMedicine 2019, 42, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Neznanov, N.; Neznanova, L.; Kondratov, R.V.; O’Rourke, D.M.; Ullrich, A.; Gudkov, A.V. The ability of protein tyrosine phosphatase SHP-1 to suppress NFκB can be inhibited by dominant negative mutant of SIRPα. DNA Cell Biol. 2004, 23, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xia, J.; Li, T.; Zhou, H.; Ouyang, W.; Hong, Z.; Ke, Y.; Qian, J.; Xu, F. Shp2 Deficiency impairs the inflammatory response againsthaemophilus influenzaeby regulating macrophage polarization. J. Infect. Dis. 2016, 214, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Alsadeq, A.; Hobeika, E.; Medgyesi, D.; Kläsener, K.; Reth, M. The role of the Syk/Shp-1 kinase-phosphatase equilibrium in B cell development and signaling. J. Immunol. 2014, 193, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.-Y.; Hunter, S.; Kim, M.-K.; Indik, Z.K.; Schreiber, A.D. The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcγ receptors FcγRIIB and FcγRIIA. J. Leukoc. Biol. 2003, 73, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, J.; Rodríguez, E.; Van Kooyk, Y. Modulation of immune tolerance via Siglec-Sialic acid interactions. Front. Immunol. 2018, 9, 2807. [Google Scholar] [CrossRef] [Green Version]
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- Cesano, A.; Gayko, U. CD22 as a target of passive immunotherapy. Semin. Oncol. 2003, 30, 253–257. [Google Scholar] [CrossRef]
- Suresh, T.; Lee, L.X.; Joshi, J.; Barta, S.K. New antibody approaches to lymphoma therapy. J. Hematol. Oncol. 2014, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-Y.; Weiner, G. Complement and cellular cytotoxicity in antibody therapy of cancer. Expert Opin. Biol. Ther. 2008, 8, 759–768. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Dörken, B.; Moldenhauer, G.; Pezzutto, A.; Schwartz, R.; Feller, A.; Kiesel, S.; Nadler, L.M. HD39 (B3), a B lineage-restricted antigen whose cell surface expression is limited to resting and activated human B lymphocytes. J. Immunol. 1986, 136, 4470–4479. [Google Scholar]
- Leonard, J.P.; Coleman, M.; Ketas, J.C.; Chadburn, A.; Furman, R.; Schuster, M.W.; Feldman, E.J.; Ashe, M.; Schuster, S.J.; Wegener, W.A.; et al. Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin’s lymphoma. Clin. Cancer Res. 2004, 10, 5327–5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, J.P.; Goldenberg, D.M. Preclinical and clinical evaluation of epratuzumab (anti-CD22 IgG) in B-cell malignancies. Oncogene 2007, 26, 3704–3713. [Google Scholar] [CrossRef] [Green Version]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, A potent and selective anti-CD33 antibody−calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjugate Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-Cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Sievers, E.; Appelbaum, F.; Spielberger, R.; Forman, S.; Flowers, D.; Smith, F.; Shannon-Dorcy, K.; Berger, M.; Bernstein, I. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: A phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, F.R.; Bernstein, I.D. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood 2017, 130, 2373–2376. [Google Scholar] [CrossRef] [Green Version]
- Thota, S.; Advani, A. Inotuzumab ozogamicin in relapsed B-cell acute lymphoblastic leukemia. Eur. J. Haematol. 2017, 98, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Bertamini, L.; Nanni, J.; Marconi, G.; Abbenante, M.; Robustelli, V.; Bacci, F.; Matti, A.; Paolini, S.; Sartor, C.; Monaco, S.L.; et al. Inotuzumab ozogamicin is effective in relapsed/refractory extramedullary B acute lymphoblastic leukemia. BMC Cancer 2018, 18, 1117. [Google Scholar] [CrossRef]
- De Vries, J.F.; Zwaan, C.M.; De Bie, M.; Voerman, J.S.A.; Boer, M.D.; Van Dongen, J.; Van Der Velden, V.H.J. The novel calicheamicin-conjugated CD22 antibody inotuzumab ozogamicin (CMC-544) effectively kills primary pediatric acute lymphoblastic leukemia cells. Leukemia 2011, 26, 255–264. [Google Scholar] [CrossRef]
- Bhojwani, D.; Sposto, R.; Shah, N.N.; Rodriguez, V.; Yuan, C.; Stetler-Stevenson, M.; O’Brien, M.M.; McNeer, J.L.; Quereshi, A.; Cabannes, A.; et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 2019, 33, 884–892. [Google Scholar] [CrossRef]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-Cell non-Hodgkin’s lymphoma: Results of a phase I study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar] [CrossRef]
- Fayad, L.; Offner, F.; Smith, M.; Verhoef, G.; Johnson, P.; Kaufman, J.L.; Rohatiner, A.; Advani, A.; Foran, J.; Hess, G.; et al. Safety and clinical activity of a combination therapy comprising two antibody-based targeting agents for the treatment of non-Hodgkin lymphoma: Results of a phase I/II study evaluating the immunoconjugate inotuzumab ozogamicin with rituximab. J. Clin. Oncol. 2013, 31, 573–583. [Google Scholar] [CrossRef]
- Dang, N.H.; Ogura, M.; Castaigne, S.; Fayad, L.E.; Jerkeman, M.; Radford, J.; Bondarenko, I.; Stewart, D.A.; Shnaidman, M.; Sullivan, S.; et al. Randomized, phase 3 trial of ino-tuzumab ozogamicin plus rituximab versus chemotherapy plus rituximab for relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Br. J. Haematol. 2018, 182, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Kreitman, R.J.; Pastan, I. Contextualizing the use of moxetumomab pasudotox in the treatment of relapsed or refractory hairy cell leukemia. Oncologist 2019, 25, e170–e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Karlin, L.; Robak, T.; Gladstone, D.E.; Le Coutre, P.; Dietrich, S.; Gotic, M.; et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018, 32, 1768–1777. [Google Scholar] [CrossRef]
- Duell, J.; Lammers, P.E.; Djuretic, I.; Chunyk, A.G.; Alekar, S.; Jacobs, I.; Gill, S. Bispecific antibodies in the treatment of hematologic malignancies. Clin. Pharmacol. Ther. 2019, 106, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Suurs, F.V.; Hooge, M.N.L.-D.; de Vries, E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Köhnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.; Zhukovsky, E.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subklewe, M.; Stein, A.S.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.S.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary results from a phase 1 first-in-human study of AMG 673, a novel half-life extended (HLE) anti-CD33/CD3 BiTE®(bispecific t-cell engager) in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Clark, M.C.; Stein, A. CD33 directed bispecific antibodies in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2020, 33, 101224. [Google Scholar] [CrossRef]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, K.M.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16x33 bispecific killer cell engager (BiKE) and ADAM17 inhibition. Clin. Cancer Res. 2014, 19, 612–626. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL-15 trispecific killer engagers (TriKEs) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin. Cancer Res. 2016, 176, 139–148. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and Trispecific killer cell engagers directly activate human NK cells through CD16 signaling and Induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [Green Version]
- Melief, C.J. Tumor eradication by adoptive transfer of cytototic T lymphocytes. Adv. Cancer Res. 1992, 58, 143–175. [Google Scholar] [CrossRef]
- Acharya, U.H.; Walter, R.B. Chimeric antigen receptor (CAR)-modified immune effector cell therapy for acute myeloid leukemia (AML). Cancers 2020, 12, 3617. [Google Scholar] [CrossRef] [PubMed]
- Masoumi, J.; Jafarzadeh, A.; Abdolalizadeh, J.; Khan, H.; Philippe, J.; Mirzaei, H.; Mirzaei, H.R. Cancer stem cell-targeted chimeric antigen receptor (CAR)-T cell therapy: Challenges and prospects. Acta Pharm. Sin. B 2020, 11, 1721–1739. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 2019, 33, 2854–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Deng, B.; Yin, Z.; Lin, Y.; An, L.; Liu, D.; Pan, J.; Yu, X.; Chen, B.; Wu, T.; et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am. J. Hematol. 2021, 96, 671–679. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, H.; Ho, J.; Liu, R.; Wang, L.; Kuang, N.; Zheng, M.; Liu, L.; Li, J. Dual specific CD19/CD22-targeted chimeric antigen receptor T-cell therapy for refractory diffuse large B-cell lymphoma: A case report. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; Von Bonin, M.; Bejestani, E.P.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [Green Version]
- Epperly, R.; Gottschalk, S.; Velasquez, M. Harnessing T Cells to Target Pediatric Acute Myeloid Leukemia: CARs, BiTEs, and Beyond. Children 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandus, C.; Simon, H.-U.; von Gunten, S. Targeting Siglecs—A novel pharmacological strategy for immuno- and glycotherapy. Biochem. Pharmacol. 2011, 82, 323–332. [Google Scholar] [CrossRef]
- Duan, S.; Paulson, J.C. Siglecs as immune cell checkpoints in disease. Annu. Rev. Immunol. 2020, 38, 365–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, B.E.; Blixt, O.; Han, S.; Duong, B.; Li, H.; Nathan, J.K.; Bovin, N.; Paulson, J.C. High-affinity ligand probes of CD22 overcome the threshold set bycisligands to allow for binding, endocytosis, and killing of B cells. J. Immunol. 2006, 177, 2994–3003. [Google Scholar] [CrossRef] [Green Version]
- Kelm, S.; Brossmer, R.; Isecke, R.; Gross, H.-J.; Strenge, K.; Schauer, R. Functional groups of sialic acids involved in binding to siglecs (sialoadhesins) deduced from interactions with synthetic analogues. JBIC J. Biol. Inorg. Chem. 1998, 255, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelm, S.; Pelz, A.; Schauer, R.; Filbin, M.T.; Tang, S.; de Bellard, M.E.; Schnaar, R.; Mahoney, J.A.; Hartnell, A.; Bradfield, P.; et al. Sialoadhesin, myelin-associated glycoprotein and CD22 define a new family of sialic acid-dependent adhesion molecules of the immunoglobulin superfamily. Curr. Biol. 1994, 4, 965–972. [Google Scholar] [CrossRef]
- Collins, B.E.; Blixt, O.; DeSieno, A.R.; Bovin, N.; Marth, J.D.; Paulson, J.C. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc. Natl. Acad. Sci. USA 2004, 101, 6104–6109. [Google Scholar] [CrossRef] [Green Version]
- May, A.; Robinson, R.; Vinson, M.; Crocker, P.; Jones, E. Crystal structure of the N-terminal domain of sialoadhesin in complex with 3′ sialyllactose at 1.85 Å resolution. Mol. Cell 1998, 1, 719–728. [Google Scholar] [CrossRef]
- Van Rossenberg, S.M.W.; Sliedregt, L.A.J.M.; Autar, R.; Piperi, C.; Van der Merwe, A.P.; van Berkel, T.J.C.; Kuiper, J.; Biessen, E.A.L. A structure-function study of ligand recognition by CD22β. J. Biol. Chem. 2001, 276, 12967–12973. [Google Scholar] [CrossRef] [Green Version]
- Kelm, S.; Gerlach, J.; Brossmer, R.; Danzer, C.-P.; Nitschke, L. The ligand-binding domain of CD22 is needed for inhibition of the B cell receptor signal, as demonstrated by a novel human CD22-specific inhibitor compound. J. Exp. Med. 2002, 195, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- Zaccai, N.R.; Maenaka, K.; Maenaka, T.; Crocker, P.; Brossmer, R.; Kelm, S.; Jones, E. Structure-guided design of sialic acid-based siglec inhibitors and crystallographic analysis in complex with sialoadhesin. Structure 2003, 11, 557–567. [Google Scholar] [CrossRef]
- Chabre, Y.M.; Roy, R. Design and creativity in synthesis of multivalent neoglycoconjugates. Adv. Carbohyd. Chem. Biochem. 2010, 63, 165–393. [Google Scholar]
- Chen, W.C.; Sigal, D.S.; Saven, A.; Paulson, J.C. Targeting B lymphoma with nanoparticles bearing glycan ligands of CD22. Leuk. Lymphoma 2011, 53, 208–210. [Google Scholar] [CrossRef]
- Chen, W.C.; Completo, G.C.; Sigal, D.S. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood 2011, 117, 5551. [Google Scholar] [CrossRef] [Green Version]
- Makwana, V.; Karanjia, J.; Haselhorst, T.; Anoopkumar-Dukie, S.; Rudrawar, S. Liposomal doxorubicin as targeted delivery platform: Current trends in surface functionalization. Int. J. Pharm. 2021, 593, 120117. [Google Scholar] [CrossRef]
- O’Reilly, M.K.; Tian, H.; Paulson, J.C. CD22 Is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B Cells. J. Immunol. 2010, 186, 1554–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, Z.; Zhang, T.; Wang, X.; Li, X.; Song, Y.; Cheng, X.; Huang, Z.; Deng, Y. The anticancer efficacy of pixantrone-loaded liposomes decorated with sialic acid–octadecylamine conjugate. Biomaterials 2014, 35, 5216–5225. [Google Scholar] [CrossRef]
- Zhou, S.; Zhang, T.; Peng, B.; Luo, X.; Liu, X.; Hu, L.; Liu, Y.; Di, D.; Song, Y.; Deng, Y. Targeted delivery of epirubicin to tumor-associated macrophages by sialic acid-cholesterol conjugate modified liposomes with improved antitumor activity. Int. J. Pharm. 2017, 523, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhao, D.; Hu, Y.; Liu, M.; Liao, X.; Zhao, B.; Liu, X.; Deng, Y.; Song, Y. Terminating the renewal of tumor-associated macrophages: A sialic acid-based targeted delivery strategy for cancer immunotherapy. Int. J. Pharm. 2019, 571, 118706. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Yu, C.; Wang, P.; Shi, Y.; Cao, W.; Cheng, B.; Chapla, D.G.; Ma, Y.; Li, J.; Rodrigues, E.; et al. Glycoengineering of NK Cells with Glycan Ligands of CD22 and Selectins for B-Cell Lymphoma Therapy. Angew. Chem. Int. Ed. 2021, 60, 3603–3610. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Läubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, J.; Sari-Ak, D.; Bagga, T. Siglecs as Therapeutic Targets in Cancer. Biology 2021, 10, 1178. https://doi.org/10.3390/biology10111178
Lim J, Sari-Ak D, Bagga T. Siglecs as Therapeutic Targets in Cancer. Biology. 2021; 10(11):1178. https://doi.org/10.3390/biology10111178
Chicago/Turabian StyleLim, Jackwee, Duygu Sari-Ak, and Tanaya Bagga. 2021. "Siglecs as Therapeutic Targets in Cancer" Biology 10, no. 11: 1178. https://doi.org/10.3390/biology10111178
APA StyleLim, J., Sari-Ak, D., & Bagga, T. (2021). Siglecs as Therapeutic Targets in Cancer. Biology, 10(11), 1178. https://doi.org/10.3390/biology10111178