
Pressure Adaptations in Deep-Sea Moritella Dihydrofolate Reductases: Compressibility versus Stability


Abstract
:Simple Summary
Abstract
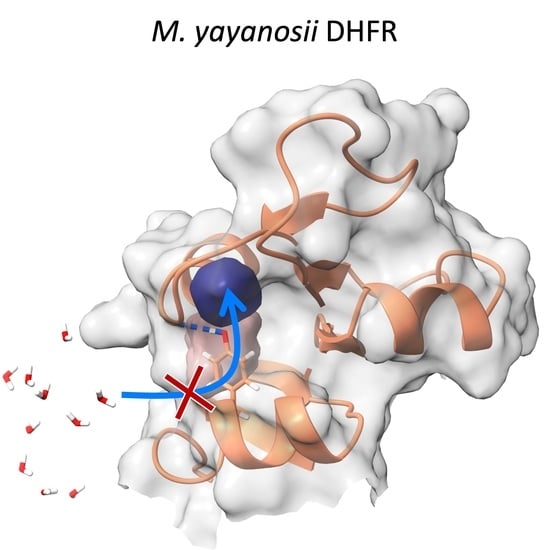
1. Introduction
2. Materials and Methods
3. Results
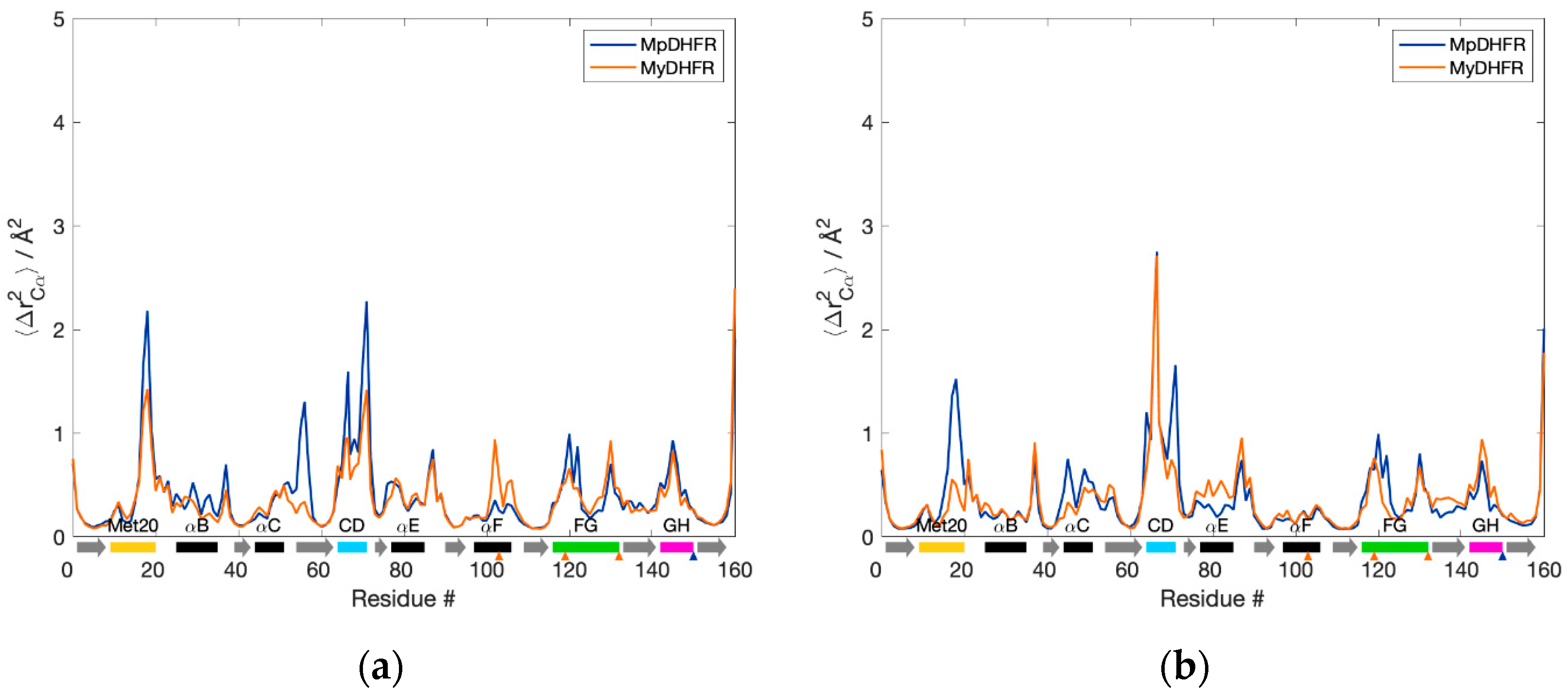
3.1. Average Properties
3.2. Hydrogen Bonding
3.3. Potential Energy Landscape
3.4. Cavities and Clefts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ichiye, T. What makes proteins work: Exploring life in P-T-X. Phys. Biol. 2016, 13, 063001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somero, G.N. Proteins and temperature. Annu. Rev. Physiol. 1995, 57, 453–468. [Google Scholar] [CrossRef]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol. 2003, 1, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Jebbar, M.; Franzetti, B.; Girard, E.; Oger, P. Microbial diversity and adaptation to high hydrostatic pressure in deep-sea hydrothermal vents prokaryotes. Extremophiles 2015, 19, 721–740. [Google Scholar] [CrossRef] [PubMed]
- Kallmeyer, J.; Pockalny, R.; Adhikari, R.R.; Smith, D.C.; D’Hondt, S. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. USA 2012, 109, 16213–16216. [Google Scholar] [CrossRef] [Green Version]
- Yayanos, A.A. Deep-sea piezophilic bacteria. Methods Microbiol. 2001, 30, 615–637. [Google Scholar]
- Bartlett, D.; Wright, M.; Yayanos, A.A.; Silverman, M. Isolation of a gene regulated by hydrostatic-pressure in a deep-sea bacterium. Nature 1989, 342, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Jaenicke, R. Review: Proteins under pressure: The influence of high hydrostatic pressure on structure, function and assembly of proteins and protein complexes. Eur. J. Biochem. 1994, 221, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Weber, G. Pressure stability of proteins. Annu. Rev. Phys. Chem. 1993, 44, 89–113. [Google Scholar] [CrossRef]
- Roche, J.; Caro, J.A.; Norberto, D.R.; Barthe, P.; Roumestand, C.; Schlessman, J.L.; Garcia, A.E.; Garcia-Moreno, B.E.; Royer, C.A. Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 6945–6950. [Google Scholar] [CrossRef] [Green Version]
- Frye, K.J.; Royer, C.A. Probing the contribution of internal cavities to the volume change of protein unfolding under pressure. Protein Sci. 1998, 7, 2217–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitahara, R.; Hata, K.; Maeno, A.; Akasaka, K.; Chimenti, M.S.; Garcia-Moreno, B.; Schroer, M.A.; Jeworrek, C.; Tolan, M.; Winter, R.; et al. Structural plasticity of staphylococcal nuclease probed by perturbation with pressure and pH. Proteins 2011, 79, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.D.; Quillin, M.L.; Hummer, G.; Matthews, B.W.; Gruner, S.M. Structural rigidity of a large cavity-containing protein revealed by high-pressure crystallography. J. Mol. Biol. 2007, 367, 752–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagae, T.; Kawamura, T.; Chavas, L.M.G.; Niwa, K.; Hasegawa, M.; Kato, C.; Watanabe, N. High-pressure-induced water penetration into 3-isopropylmalate dehydrogenase. Acta Crystallogr. D 2012, 68, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Nucci, N.V.; Fuglestad, B.; Athanasoula, E.A.; Wand, A.J. Role of cavities and hydration in the pressure unfolding of T-4 lysozyme. Proc. Natl. Acad. Sci. USA 2014, 111, 13846–13851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagae, T.; Kato, C.; Watanabe, N. Structural analysis of 3-isopropylmalate dehydrogenase from the obligate piezophile Shewanella benthica DB21MT-2 and the nonpiezophile Shewanella oneidensis MR-1. Acta Crystallogr. F 2012, 68, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiyama, T.; Gekko, K. Effect of ligand binding on the flexibility of dihydrofolate reductase as revealed by compressibility. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1478, 257–266. [Google Scholar] [CrossRef]
- Gekko, K.; Kamiyama, T.; Ohmae, E.; Katayanagi, K. Single amino acid substitutions in flexible loops can induce large compressibility changes in dihydrofolate reductase. J. Biochem. 2000, 128, 21–27. [Google Scholar] [CrossRef]
- Wan, Q.; Bennett, B.C.; Wilson, M.A.; Kovalevsky, A.; Langan, P.; Howell, E.E.; Dealwis, C. Toward resolving the catalytic mechanism of dihydrofolate reductase using neutron and ultrahigh-resolution X-ray crystallography. Proc. Natl. Acad. Sci. USA 2014, 111, 18225–18230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.T.; Francis, K.; Layfield, J.P.; Huang, X.Y.; Hammes-Schiffer, S.; Kohen, A.; Benkovic, S.J. Escherichia coli dihydrofolate reductase catalyzed proton and hydride transfers: Temporal order and the roles of Asp27 and Tyr100. Proc. Natl. Acad. Sci. USA 2014, 111, 18231–18236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef]
- Epstein, D.M.; Benkovic, S.J.; Wright, P.E. Dynamics of the dihydrofolate reductase-folate complex-catalytic sites and regions known to undergo conformational change exhibit diverse dynamical features. Biochemistry 1995, 34, 11037–11048. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [Google Scholar] [CrossRef]
- Bhabha, G.; Ekiert, D.C.; Jennewein, M.; Zmasek, C.M.; Tuttle, L.M.; Kroon, G.; Dyson, H.J.; Godzik, A.; Wilson, I.A.; Wright, P.E. Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1262. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Nogi, Y.; Kato, C.; Liang, Z.; Rüger, H.-J.; de Kegel, D.; Glansdorff, N. Moritella profunda sp. nov. and Moritella abyssi sp. nov., two psychropiezophilic organisms isolated from deep Atlantic sediments. Int. J. Syst. Evol. Microbiol. 2003, 53, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Hata, K.; Kono, R.; Fujisawa, M.; Kitahara, R.; Kamatari, Y.O.; Akasaka, K.; Xu, Y. High pressure NMR study of dihydrofolate reductase from a deep-sea bacterium Moritella profunda. Cell Mol. Biol. 2004, 50, 311–316. [Google Scholar]
- Ohmae, E.; Murakami, C.; Tate, S.-I.; Gekko, K.; Hata, K.; Akasaka, K.; Kato, C. Pressure dependence of activity and stability of dihydrofolate reductases of the deep-sea bacterium Moritella profunda and Escherichia coli. Biochim. Biophys. Acta 2012, 1824, 511–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmae, E.; Miyashita, Y.; Tate, S.; Gekko, K.; Kitazawa, S.; Kitahara, R.; Kuwajima, K. Solvent environments significantly affect the enzymatic function of Escherichia coli dihydrofolate reductase: Comparison of wild-type protein and active-site mutant D27E. Biochim. Biophys. Acta 2013, 1834, 2782–2794. [Google Scholar] [CrossRef]
- Ohmae, E.; Kubota, K.; Nakasone, K.; Kato, C.; Gekko, K. Pressure-dependent activity of dihydrofolate reductase from a deep-sea bacterium Shewanella violacea strain DSS12. Chem. Lett. 2004, 33, 798–799. [Google Scholar] [CrossRef]
- Murakami, C.; Ohmae, E.; Tate, S.-I.; Gekko, K.; Nakasone, K.; Kato, C. Cloning and characterization of dihydrofolate reductases from deep-sea bacteria. J. Biochem. 2010, 147, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Murakami, C.; Ohmae, E.; Tate, S.-I.; Gekko, K.; Nakasone, K.; Kato, C. Comparative study on dihydrofolate reductases from Shewanella species living in deep-sea and ambient atmospheric-pressure environments. Extremophiles 2010, 15, 165–175. [Google Scholar] [CrossRef]
- Ichiye, T. Enzymes from piezophiles. Semin. Cell Dev. Biol. 2018, 84, 138–146. [Google Scholar] [CrossRef]
- Ohmae, E.; Miyashita, Y.; Kato, C. Thermodynamic and functional characteristics of deep-sea enzymes revealed by pressure effects. Extremophiles 2013, 17, 701–709. [Google Scholar] [CrossRef]
- Evans, R.M.; Behiry, E.M.; Tey, L.-H.; Guo, J.; Loveridge, E.J.; Allemann, R.K. Catalysis by dihydrofolate reductase from the psychropiezophile Moritella profunda. ChemBioChem 2010, 11, 2010–2017. [Google Scholar] [CrossRef]
- Huang, Q.; Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Extreme biophysics: Enzymes under pressure. J. Comput. Chem. 2017, 38, 1174–1182. [Google Scholar] [CrossRef]
- Huang, Q.; Tran, K.N.; Rodgers, J.M.; Bartlett, D.H.; Hemley, R.J.; Ichiye, T. A molecular perspective on the limits of life: Enzymes under pressure. Condens. Matter Phys. 2016, 19, 22801–22817. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Adaptations for pressure and temperature effects on loop motion in Escherichia coli and Moritella profunda dihydrofolate reductase. High Press. Res. 2019, 39, 225–237. [Google Scholar] [CrossRef]
- Penhallurick, R.W.; Harold, A.; Durnal, M.D.; Ichiye, T. How adding a single methylene to dihydrofolate reductase can change its conformational dynamics. J. Chem. Phys. 2021, 154, 165103. [Google Scholar] [CrossRef]
- Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Quasiharmonic analysis of protein energy landscapes from pressure-temperature molecular dynamics simulations. J. Chem. Phys. 2017, 147, 125103–125110. [Google Scholar] [CrossRef]
- Huang, Q.; Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Quasiharmonic Analysis of the Energy Landscapes of Dihydrofolate Reductase from Piezophiles and Mesophiles. J. Phys. Chem. B 2018, 122, 5527–5533. [Google Scholar] [CrossRef]
- Penhallurick, R.W.; Durnal, M.D.; Harold, A.; Ichiye, T. Adaptations for Pressure and Temperature in Dihydrofolate Reductases. Microorganisms 2021, 9, 1706. [Google Scholar] [CrossRef]
- Nogi, Y.; Kato, C. Taxonomic studies of extremely barophilic bacteria isolated from the Mariana Trench and description of Moritella yayanosii sp. nov., a new barophilic bacterial isolate. Extremophiles 1999, 3, 71–77. [Google Scholar]
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Coutsias, E.A.; Seok, C.; Jacobson, M.P.; Dill, K.A. A kinematic view of loop closure. J. Comput. Chem. 2004, 25, 510–528. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L.; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellot, M.; Dunbrack, R.L., Jr.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph, D.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Pavelites, J.J.; Gao, J.L.; Bash, P.A.; Mackerell, A.D. A molecular mechanics force field for NAD(+), NADH; the pyrophosphate groups of nucleotides. J. Comput. Chem. 1997, 18, 221–239. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Liu, D.C.; Nocedal, J. On the Limited Memory BFGS Method for Large-Scale Optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.C. Molecular-dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Aqvist, J.; Wennerstrom, P.; Nervall, M.; Bjelic, S.; Brandsdal, B.O. Molecular dynamics simulations of water and biomolecules wit a Monte Carlo constant pressure algorithm. Chem. Phys. Lett. 2004, 384, 288–294. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nose-Hoover Chains—The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- De Loof, H.; Nilsson, L.; Rigler, R. Molecular dynamics simulation of galanin in aqueous and nonaqueous solution. J. Am. Chem. Soc. 1992, 114, 4028–4035. [Google Scholar] [CrossRef]
- Till, M.S.; Ullmann, G.M. McVol—A program for calculating protein volumes and identifying cavities by a Monte Carlo algorithm. J. Mol. Model. 2010, 16, 419–429. [Google Scholar] [CrossRef]
- Keedy, D.A.; van den Bedem, H.; Sivak, D.A.; Petsko, G.A.; Ringe, D.; Wilson, M.A.; Fraser, J.S. Crystal Cryocooling Distorts Conformational Heterogeneity in a Model Michaelis Complex of DHFR. Structure 2014, 22, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Fenwick, R.B.; van den Bedem, H.; Fraser, J.S.; Wright, P.E. Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proc. Natl. Acad. Sci. USA 2014, 111, E445–E454. [Google Scholar] [CrossRef] [Green Version]
- Nagae, T.; Yamada, H.; Watanabe, N. High-pressure protein crystal structure analysis of Escherichia coli dihydrofolate reductase complexed with folate and NADP+. Acta Crystallogr. D 2018, 74, 895–905. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | P (bar) | 〈ΔrHA2〉 (Å2) | NHB | 〈Rg〉 (Å2) |
|---|---|---|---|---|
| MpDHFR | 1 | 0.60 ± 0.04 | 104 ± 1 | 15.48 ± 0.07 |
| MpDHFR | 800 | 0.61 ± 0.02 | 106 ± 3 | 15.50 ± 0.04 |
| MyDHFR | 1 | 0.60 ± 0.06 | 107 ± 1 | 15.63 ± 0.03 |
| MyDHFR | 800 | 0.61 ± 0.08 | 105 ± 1 | 15.56 ± 0.05 |
| DHFR | σ02 (Å2) | 𝛼P,0 (10−3/K) | 𝜅T,0 (10−3/kbar) | 𝜒2 (10−6) | Tg,0 (K) | –c (K/kbar) | 𝜒2 (10−6) |
|---|---|---|---|---|---|---|---|
| MpDHFR | 0.148 ± 0.001 | 8.0 ± 0.1 | 65 ± 2 | 3.5 | 191 ± 2 | 0.5 ± 0.3 | 0.51 |
| MyDHFR | 0.151 ± 0.001 | 8.1 ± 0.1 | 70 ± 3 | 4.0 | 186 ± 1 | 1.0 ± 0.2 | 0.39 |
| DHFR | P (bar) | Cavity/Cleft | |||||
|---|---|---|---|---|---|---|---|
| 1 | Cavity 2 | Cleft 2 | 3 | 4 | 5 | ||
| MpDHFR | 1 | 17 ± 8 | 1 ± 3 | 5 ± 7 | 19 ± 5 | 12 ± 9 | 4 ± 5 |
| MpDHFR | 800 | 15 ± 5 | 1 ± 0 | 3 ± 8 | 19 ± 3 | 11 ± 6 | 2 ± 3 |
| MyDHFR | 1 | 16 ± 6 | 0 ± 0 | 10 ± 13 | 19 ± 5 | 10 ± 12 | 8 ± 5 |
| MyDHFR | 800 | 16 ± 3 | 2 ± 5 | 8 ± 8 | 19 ± 5 | 15 ± 17 | 2 ± 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penhallurick, R.W.; Ichiye, T. Pressure Adaptations in Deep-Sea Moritella Dihydrofolate Reductases: Compressibility versus Stability. Biology 2021, 10, 1211. https://doi.org/10.3390/biology10111211
Penhallurick RW, Ichiye T. Pressure Adaptations in Deep-Sea Moritella Dihydrofolate Reductases: Compressibility versus Stability. Biology. 2021; 10(11):1211. https://doi.org/10.3390/biology10111211
Chicago/Turabian StylePenhallurick, Ryan W., and Toshiko Ichiye. 2021. "Pressure Adaptations in Deep-Sea Moritella Dihydrofolate Reductases: Compressibility versus Stability" Biology 10, no. 11: 1211. https://doi.org/10.3390/biology10111211