δ-Cells: The Neighborhood Watch in the Islet Community


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
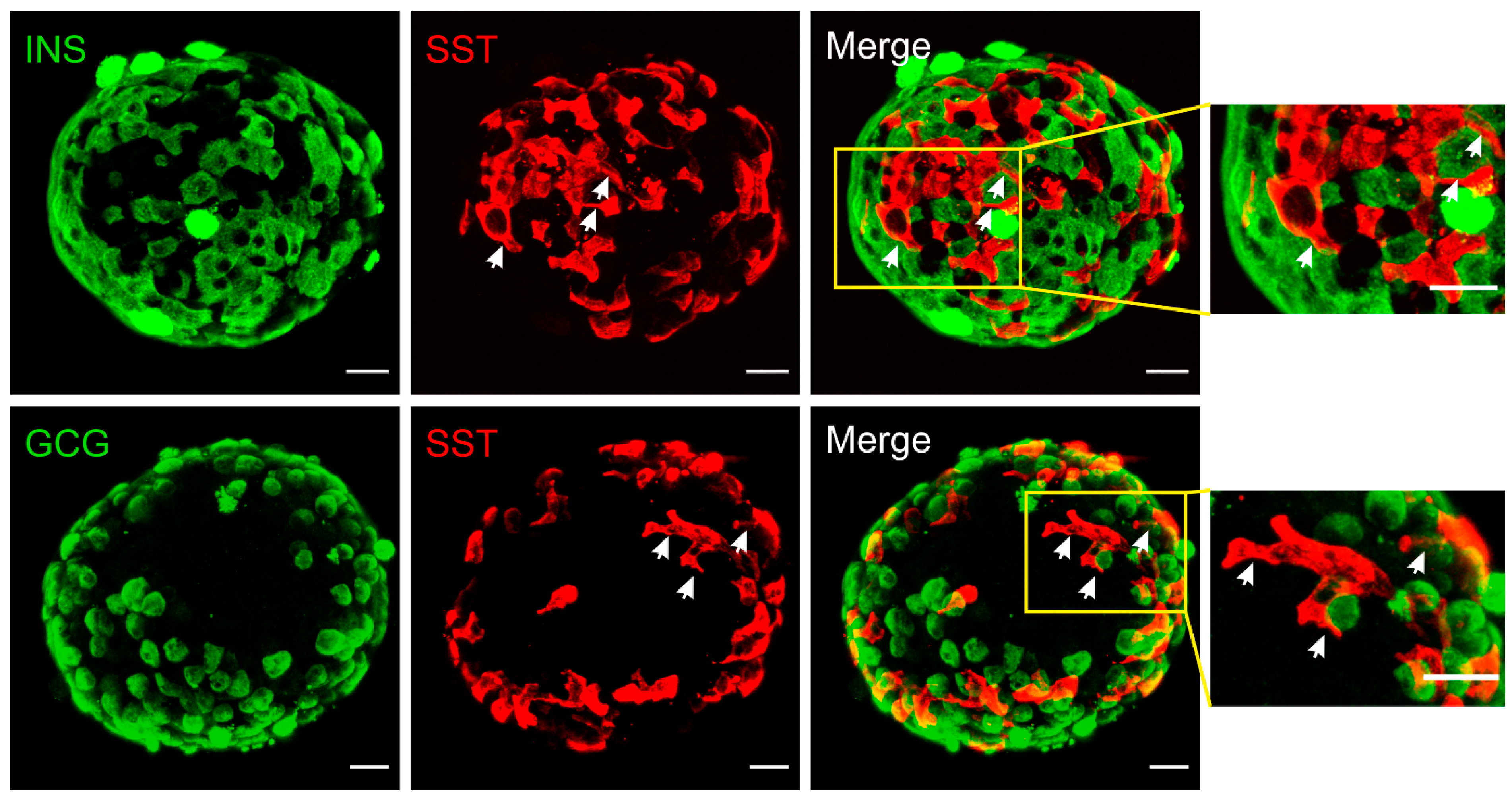
2. Structural Basis for δ-Cell Paracrine Regulation
3. Nutritional Regulation of Somatostatin Secretion
3.1. Glucose
3.2. Fatty Acids
3.3. Amino Acids
4. β-Cell-Mediated Paracrine Regulation of δ-Cells
4.1. Insulin
4.2. Urocortin3
4.3. Gap Junction
4.4. γ-Aminobutyric Acid
4.5. Serotonin
5. α-Cell-Mediated Paracrine Regulation of δ-Cells
5.1. Glucagon
5.2. Glutamate
5.3. Glucagon-Like Peptide-1
5.4. Acetylcholine
5.5. Glicentin-Related Pancreatic Polypeptide
6. δ-Cell-Mediated Intra-Islet Coordination
6.1. Somatostatin
6.2. Neuronostatin
6.3. Cortistatin
7. Paracrine Crosstalk of δ-Cells in Diabetes
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brissova, M.; Fowler, M.J.; Nicholson, W.E.; Chu, A.; Hirshberg, B.; Harlan, D.M.; Powers, A.C. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 2005, 53, 1087–1097. [Google Scholar] [CrossRef]
- Rorsman, P.; Huising, M.O. The somatostatin-secreting pancreatic delta-cell in health and disease. Nat. Rev. Endocrinol. 2018, 14, 404–414. [Google Scholar] [CrossRef]
- Strowski, M.Z.; Parmar, R.M.; Blake, A.D.; Schaeffer, J.M. Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: An in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology 2000, 141, 111–117. [Google Scholar] [CrossRef]
- Omar-Hmeadi, M.; Lund, P.E.; Gandasi, N.R.; Tengholm, A.; Barg, S. Paracrine control of alpha-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat. Commun. 2020, 11, 1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brereton, M.F.; Vergari, E.; Zhang, Q.; Clark, A. Alpha-, Delta- and PP-cells: Are They the Architectural Cornerstones of Islet Structure and Co-ordination? J. Histochem. Cytochem. 2015, 63, 575–591. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Yang, Z.; Li, Q.; Yu, Z.; Chen, X.; Li, J.C.; Li, B.; Ning, S.L.; Cui, M.; Sun, J.P.; et al. Ablation of somatostatin cells leads to impaired pancreatic islet function and neonatal death in rodents. Cell Death Dis. 2018, 9, 682. [Google Scholar] [CrossRef] [Green Version]
- Adriaenssens, A.E.; Svendsen, B.; Lam, B.Y.; Yeo, G.S.; Holst, J.J.; Reimann, F.; Gribble, F.M. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 2016, 59, 2156–2165. [Google Scholar] [CrossRef] [Green Version]
- DiGruccio, M.R.; Mawla, A.M.; Donaldson, C.J.; Noguchi, G.M.; Vaughan, J.; Cowing-Zitron, C.; van der Meulen, T.; Huising, M.O. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 2016, 5, 449–458. [Google Scholar] [CrossRef] [PubMed]
- van der Meulen, T.; Donaldson, C.J.; Caceres, E.; Hunter, A.E.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, J.T.; Burdett, E.; Coy, D.H.; Giacca, A.; Efendic, S.; Vranic, M. Somatostatin receptor type 2 antagonism improves glucagon and corticosterone counterregulatory responses to hypoglycemia in streptozotocin-induced diabetic rats. Diabetes 2012, 61, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimian, N.; Qin, T.; Liang, T.; Osundiji, M.; Huang, Y.; Teich, T.; Riddell, M.C.; Cattral, M.S.; Coy, D.H.; Vranic, M.; et al. Somatostatin receptor type 2 antagonism improves glucagon counterregulation in biobreeding diabetic rats. Diabetes 2013, 62, 2968–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef] [Green Version]
- Arrojo, E.D.R.; Jacob, S.; Garcia-Prieto, C.F.; Zheng, X.; Fukuda, M.; Nhu, H.T.T.; Stelmashenko, O.; Pecanha, F.L.M.; Rodriguez-Diaz, R.; Bushong, E.; et al. Structural basis for delta cell paracrine regulation in pancreatic islets. Nat. Commun. 2019, 10, 3700. [Google Scholar] [CrossRef] [PubMed]
- Huising, M.O.; van der Meulen, T.; Huang, J.L.; Pourhosseinzadeh, M.S.; Noguchi, G.M. The Difference delta-Cells Make in Glucose Control. Physiology 2018, 33, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cui, M.; Yang, F.; Li, N.; Jiang, B.; Yu, Z.; Zhang, D.; Wang, Y.; Zhu, X.; Hu, H.; et al. A cullin 4B-RING E3 ligase complex fine-tunes pancreatic delta cell paracrine interactions. J. Clin. Investig. 2017, 127, 2631–2646. [Google Scholar] [CrossRef] [Green Version]
- Briant, L.J.B.; Reinbothe, T.M.; Spiliotis, I.; Miranda, C.; Rodriguez, B.; Rorsman, P. δ-cells and β-cells are electrically coupled and regulate alpha-cell activity via somatostatin. J. Physiol. 2018, 596, 197–215. [Google Scholar] [CrossRef]
- Denwood, G.; Tarasov, A.; Salehi, A.; Vergari, E.; Ramracheya, R.; Takahashi, H.; Nikolaev, V.O.; Seino, S.; Gribble, F.; Reimann, F.; et al. Glucose stimulates somatostatin secretion in pancreatic delta-cells by cAMP-dependent intracellular Ca(2+) release. J. Gen. Physiol. 2019, 151, 1094–1115. [Google Scholar] [CrossRef] [Green Version]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Asterholm, I.W.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139. [Google Scholar] [CrossRef]
- Gopel, S.O.; Kanno, T.; Barg, S.; Rorsman, P. Patch-clamp characterisation of somatostatin-secreting -cells in intact mouse pancreatic islets. J. Physiol. 2000, 528, 497–507. [Google Scholar] [CrossRef]
- Braun, M.; Ramracheya, R.; Amisten, S.; Bengtsson, M.; Moritoh, Y.; Zhang, Q.; Johnson, P.R.; Rorsman, P. Somatostatin release, electrical activity, membrane currents and exocytosis in human pancreatic delta cells. Diabetologia 2009, 52, 1566–1578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bengtsson, M.; Partridge, C.; Salehi, A.; Braun, M.; Cox, R.; Eliasson, L.; Johnson, P.R.; Renstrom, E.; Schneider, T.; et al. R-type Ca(2+)-channel-evoked CICR regulates glucose-induced somatostatin secretion. Nat. Cell. Biol. 2007, 9, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Vergari, E.; Denwood, G.; Salehi, A.; Zhang, Q.; Adam, J.; Alrifaiy, A. Somatostatin secretion by Na+-dependent Ca2+-induced Ca2+ release in pancreatic delta cells. Nat. Metab. 2020, 2, 32–40. [Google Scholar] [CrossRef]
- Olofsson, C.S.; Salehi, A.; Gopel, S.O.; Holm, C.; Rorsman, P. Palmitate stimulation of glucagon secretion in mouse pancreatic alpha-cells results from activation of L-type calcium channels and elevation of cytoplasmic calcium. Diabetes 2004, 53, 2836–2843. [Google Scholar] [CrossRef] [Green Version]
- Stone, V.M.; Dhayal, S.; Brocklehurst, K.J.; Lenaghan, C.; Sorhede Winzell, M.; Hammar, M.; Xu, X.; Smith, D.M.; Morgan, N.G. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia 2014, 57, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Olofsson, C.S.; Salehi, A.; Holm, C.; Rorsman, P. Palmitate increases L-type Ca2+ currents and the size of the readily releasable granule pool in mouse pancreatic beta-cells. J. Physiol. 2004, 557, 935–948. [Google Scholar] [CrossRef]
- Gerber, P.P.; Trimble, E.R.; Wollheim, C.B.; Renold, A.E. Effect of insulin on glucose- and arginine-stimulated somatostatin secretion from the isolated perfused rat pancreas. Endocrinology 1981, 109, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Ipp, E.; Dobbs, R.E.; Arimura, A.; Vale, W.; Harris, V.; Unger, R.H. Release of immunoreactive somatostatin from the pancreas in response to glucose, amino acids, pancreozymin-cholecystokinin, and tolbutamide. J. Clin. Investig. 1977, 60, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Patton, G.S.; Ipp, E.; Dobbs, R.E.; Orci, L.; Vale, W.; Unger, R.H. Pancreatic immunoreactive somatostatin release. Proc. Natl. Acad. Sci. USA 1977, 74, 2140–2143. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Gaudel, C.; McClenaghan, N.H. Nutrient regulation of insulin secretion and beta-cell functional integrity. Adv. Exp. Med. Biol. 2010, 654, 91–114. [Google Scholar] [CrossRef]
- Hauge-Evans, A.C.; Anderson, R.L.; Persaud, S.J.; Jones, P.M. Delta cell secretory responses to insulin secretagogues are not mediated indirectly by insulin. Diabetologia 2012, 55, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Taniguchi, H.; Tamagawa, M.; Ejiri, K.; Baba, S. Modulation of somatostatin release by endogenous glucagon and insulin: Physiological relationship between A, B and D cells in rat pancreatic islets. Endocrinol. Jpn. 1982, 29, 503–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honey, R.N.; Fallon, M.B.; Weir, G.C. Effects of exogenous insulin, glucagon, and somatostatin on islet hormone secretion in the perfused chicken pancreas. Metabolism 1980, 29, 1242–1246. [Google Scholar] [CrossRef]
- Stagner, J.; Samols, E.; Polonsky, K.; Pugh, W. Lack of direct inhibition of insulin secretion by exogenous insulin in the canine pancreas. J. Clin. Investig. 1986, 78, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samols, E.; Stagner, J.I.; Ewart, R.B.; Marks, V. The order of islet microvascular cellular perfusion is B----A----D in the perfused rat pancreas. J. Clin. Investig. 1988, 82, 350–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunicardi, F.C.; Kleinman, R.; Moldovan, S.; Nguyen, T.H.; Watt, P.C.; Walsh, J.; Gingerich, R. Immunoneutralization of somatostatin, insulin, and glucagon causes alterations in islet cell secretion in the isolated perfused human pancreas. Pancreas 2001, 23, 302–308. [Google Scholar] [CrossRef]
- Li, C.; Chen, P.; Vaughan, J.; Lee, K.F.; Vale, W. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc. Natl. Acad. Sci. USA 2007, 104, 4206–4211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; McKenna, L.B.; Bogue, C.W.; Kaestner, K.H. The diabetes gene Hhex maintains delta-cell differentiation and islet function. Genes Dev. 2014, 28, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Ravier, M.A.; Guldenagel, M.; Charollais, A.; Gjinovci, A.; Caille, D.; Sohl, G.; Wollheim, C.B.; Willecke, K.; Henquin, J.C.; Meda, P. Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 2005, 54, 1798–1807. [Google Scholar] [CrossRef] [Green Version]
- Benninger, R.K.; Zhang, M.; Head, W.S.; Satin, L.S.; Piston, D.W. Gap junction coupling and calcium waves in the pancreatic islet. Biophys. J. 2008, 95, 5048–5061. [Google Scholar] [CrossRef] [Green Version]
- Head, W.S.; Orseth, M.L.; Nunemaker, C.S.; Satin, L.S.; Piston, D.W.; Benninger, R.K. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 2012, 61, 1700–1707. [Google Scholar] [CrossRef] [Green Version]
- Hellman, B.; Salehi, A.; Gylfe, E.; Dansk, H.; Grapengiesser, E. Glucose generates coincident insulin and somatostatin pulses and antisynchronous glucagon pulses from human pancreatic islets. Endocrinology 2009, 150, 5334–5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, G.M.; Huising, M.O. Integrating the inputs that shape pancreatic islet hormone release. Nat. Metab. 2019, 1, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi, A.; Qader, S.S.; Grapengiesser, E.; Hellman, B. Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regul. Pept. 2007, 144, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Reetz, A.; Solimena, M.; Matteoli, M.; Folli, F.; Takei, K.; De Camilli, P. GABA and pancreatic beta-cells: Colocalization of glutamic acid decarboxylase (GAD) and GABA with synaptic-like microvesicles suggests their role in GABA storage and secretion. EMBO J. 1991, 10, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Sorenson, R.L.; Garry, D.G.; Brelje, T.C. Structural and functional considerations of GABA in islets of Langerhans. Beta-cells and nerves. Diabetes 1991, 40, 1365–1374. [Google Scholar] [CrossRef]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Clark, A.; Walker, J.N.; Johnson, P.R.; Rorsman, P. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 2010, 59, 1694–1701. [Google Scholar] [CrossRef] [Green Version]
- Wendt, A.; Birnir, B.; Buschard, K.; Gromada, J.; Salehi, A.; Sewing, S.; Rorsman, P.; Braun, M. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes 2004, 53, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Bailey, S.J.; Ravier, M.A.; Rutter, G.A. Glucose-dependent regulation of gamma-aminobutyric acid (GABA A) receptor expression in mouse pancreatic islet alpha-cells. Diabetes 2007, 56, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Bennet, H.; Mollet, I.G.; Balhuizen, A.; Medina, A.; Nagorny, C.; Bagge, A.; Fadista, J.; Ottosson-Laakso, E.; Vikman, P.; Dekker-Nitert, M.; et al. Serotonin (5-HT) receptor 2b activation augments glucose-stimulated insulin secretion in human and mouse islets of Langerhans. Diabetologia 2016, 59, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y.; et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat. Med. 2010, 16, 804–808. [Google Scholar] [CrossRef]
- Kim, K.; Oh, C.M.; Ohara-Imaizumi, M.; Park, S.; Namkung, J.; Yadav, V.K.; Tamarina, N.A.; Roe, M.W.; Philipson, L.H.; Karsenty, G.; et al. Functional role of serotonin in insulin secretion in a diet-induced insulin-resistant state. Endocrinology 2015, 156, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Ohara-Imaizumi, M.; Kim, H.; Yoshida, M.; Fujiwara, T.; Aoyagi, K.; Toyofuku, Y.; Nakamichi, Y.; Nishiwaki, C.; Okamura, T.; Uchida, T.; et al. Serotonin regulates glucose-stimulated insulin secretion from pancreatic beta cells during pregnancy. Proc. Natl. Acad. Sci. USA 2013, 110, 19420–19425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaca, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; Berggren, P.O.; Caicedo, A. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell Rep. 2016, 17, 3281–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orci, L.; Unger, R.H. Functional subdivision of islets of Langerhans and possible role of D cells. Lancet 1975, 2, 1243–1244. [Google Scholar] [CrossRef]
- Dolais-Kitabgi, J.; Kitabgi, P.; Freychet, P. Glucose and glucagon do stimulate somatostatin release from isolated pancreatic islets. Diabetologia 1981, 21, 238. [Google Scholar] [CrossRef] [Green Version]
- Svendsen, B.; Holst, J.J. Paracrine regulation of somatostatin secretion by insulin and glucagon in mouse pancreatic islets. Diabetologia 2021, 64, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, B.; Larsen, O.; Gabe, M.B.N.; Christiansen, C.B.; Rosenkilde, M.M.; Drucker, D.J.; Holst, J.J. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep. 2018, 25, 1127–1134.e2. [Google Scholar] [CrossRef] [Green Version]
- Otter, S.; Lammert, E. Exciting Times for Pancreatic Islets: Glutamate Signaling in Endocrine Cells. Trends Endocrinol. Metab. 2016, 27, 177–188. [Google Scholar] [CrossRef]
- Takahashi, H.; Yokoi, N.; Seino, S. Glutamate as intracellular and extracellular signals in pancreatic islet functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, G.; Gross, R.; Puech, R.; Loubatieres-Mariani, M.M.; Bockaert, J. Glutamate stimulates glucagon secretion via an excitatory amino acid receptor of the AMPA subtype in rat pancreas. Eur. J. Pharmacol. 1993, 237, 45–50. [Google Scholar] [CrossRef]
- Uehara, S.; Muroyama, A.; Echigo, N.; Morimoto, R.; Otsuka, M.; Yatsushiro, S.; Moriyama, Y. Metabotropic glutamate receptor type 4 is involved in autoinhibitory cascade for glucagon secretion by alpha-cells of islet of Langerhans. Diabetes 2004, 53, 998–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, O.; Jacques-Silva, M.C.; Speier, S.; Yang, S.N.; Kohler, M.; Fachado, A.; Vieira, E.; Zierath, J.R.; Kibbey, R.; Berman, D.M.; et al. Glutamate is a positive autocrine signal for glucagon release. Cell Metab. 2008, 7, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroyama, A.; Uehara, S.; Yatsushiro, S.; Echigo, N.; Morimoto, R.; Morita, M.; Hayashi, M.; Yamamoto, A.; Koh, D.S.; Moriyama, Y. A novel variant of ionotropic glutamate receptor regulates somatostatin secretion from delta-cells of islets of Langerhans. Diabetes 2004, 53, 1743–1753. [Google Scholar] [CrossRef] [Green Version]
- Weir, G.C.; Mojsov, S.; Hendrick, G.K.; Habener, J.F. Glucagonlike peptide I (7-37) actions on endocrine pancreas. Diabetes 1989, 38, 338–342. [Google Scholar] [CrossRef]
- Gromada, J.; Holst, J.J.; Rorsman, P. Cellular regulation of islet hormone secretion by the incretin hormone glucagon-like peptide 1. Pflugers Arch. 1998, 435, 583–594. [Google Scholar] [CrossRef]
- Richards, P.; Parker, H.E.; Adriaenssens, A.E.; Hodgson, J.M.; Cork, S.C.; Trapp, S.; Gribble, F.M.; Reimann, F. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 2014, 63, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- De Marinis, Y.Z.; Salehi, A.; Ward, C.E.; Zhang, Q.; Abdulkader, F.; Bengtsson, M.; Braha, O.; Braun, M.; Ramracheya, R.; Amisten, S.; et al. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab. 2010, 11, 543–553. [Google Scholar] [CrossRef] [Green Version]
- de Heer, J.; Rasmussen, C.; Coy, D.H.; Holst, J.J. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 2008, 51, 2263–2270. [Google Scholar] [CrossRef] [Green Version]
- Orgaard, A.; Holst, J.J. The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia 2017, 60, 1731–1739. [Google Scholar] [CrossRef]
- Saponaro, C.; Gmyr, V.; Thevenet, J.; Moerman, E.; Delalleau, N.; Pasquetti, G.; Coddeville, A.; Quenon, A.; Daoudi, M.; Hubert, T.; et al. The GLP1R Agonist Liraglutide Reduces Hyperglucagonemia Induced by the SGLT2 Inhibitor Dapagliflozin via Somatostatin Release. Cell Rep. 2019, 28, 1447–1454.e4. [Google Scholar] [CrossRef] [Green Version]
- Hare, K.J.; Vilsboll, T.; Asmar, M.; Deacon, C.F.; Knop, F.K.; Holst, J.J. The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes 2010, 59, 1765–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilon, P.; Henquin, J.C. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr. Rev. 2001, 22, 565–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Diaz, R.; Dando, R.; Jacques-Silva, M.C.; Fachado, A.; Molina, J.; Abdulreda, M.H.; Ricordi, C.; Roper, S.D.; Berggren, P.O.; Caicedo, A. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat. Med. 2011, 17, 888–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honey, R.N.; Weir, G.C. Acetylcholine stimulates insulin, glucagon, and somatostatin release in the perfused chicken pancreas. Endocrinology 1980, 107, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.; Rodriguez-Diaz, R.; Fachado, A.; Jacques-Silva, M.C.; Berggren, P.O.; Caicedo, A. Control of insulin secretion by cholinergic signaling in the human pancreatic islet. Diabetes 2014, 63, 2714–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patzelt, C.; Schiltz, E. Conversion of proglucagon in pancreatic alpha cells: The major endproducts are glucagon and a single peptide, the major proglucagon fragment, that contains two glucagon-like sequences. Proc. Natl. Acad. Sci. USA 1984, 81, 5007–5011. [Google Scholar] [CrossRef] [Green Version]
- Ohneda, A.; Kobayashi, T.; Nihei, J. Effect of glicentin-related peptides on glucagon secretion in anaesthetized dogs. Diabetologia 1986, 29, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Yanaihara, C.; Matsumoto, T.; Hong, Y.M.; Yanaihara, N. Isolation and chemical characterization of glicentin C-terminal hexapeptide in porcine pancreas. FEBS Lett. 1985, 189, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Whiting, L.; Stewart, K.W.; Hay, D.L.; Harris, P.W.; Choong, Y.S.; Phillips, A.R.; Brimble, M.A.; Cooper, G.J. Glicentin-related pancreatic polypeptide inhibits glucose-stimulated insulin secretion from the isolated pancreas of adult male rats. Physiol. Rep. 2015, 3, e12638. [Google Scholar] [CrossRef] [Green Version]
- Gutniak, M.; Grill, V.; Wiechel, K.L.; Efendic, S. Basal and meal-induced somatostatin-like immunoreactivity in healthy subjects and in IDDM and totally pancreatectomized patients. Effects of acute blood glucose normalization. Diabetes 1987, 36, 802–807. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schaer, J.C.; Wenger, S.; Hoeger, C.; Erchegyi, J.; Waser, B.; Rivier, J. SST3-selective potent peptidic somatostatin receptor antagonists. Proc. Natl. Acad. Sci. USA 2000, 97, 13973–13978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivier, J.; Gulyas, J.; Kirby, D.; Low, W.; Perrin, M.H.; Kunitake, K.; DiGruccio, M.; Vaughan, J.; Reubi, J.C.; Waser, B.; et al. Potent and long-acting corticotropin releasing factor (CRF) receptor 2 selective peptide competitive antagonists. J. Med. Chem. 2002, 45, 4737–4747. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Brendel, M.D.; Zacharias, S.; Mergler, S.; Jahr, H.; Wiedenmann, B.; Bretzel, R.G.; Plockinger, U.; Strowski, M.Z. Characterization of somatostatin receptor subtype-specific regulation of insulin and glucagon secretion: An in vitro study on isolated human pancreatic islets. J. Clin. Endocrinol. Metab. 2007, 92, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirone, T.A.; Norman, M.A.; Moldovan, S.; DeMayo, F.J.; Wang, X.P.; Brunicardi, F.C. Pancreatic somatostatin inhibits insulin secretion via SSTR-5 in the isolated perfused mouse pancreas model. Pancreas 2003, 26, e67–e73. [Google Scholar] [CrossRef] [PubMed]
- Benner, C.; van der Meulen, T.; Caceres, E.; Tigyi, K.; Donaldson, C.J.; Huising, M.O. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genom. 2014, 15, 620. [Google Scholar] [CrossRef] [Green Version]
- Kailey, B.; van de Bunt, M.; Cheley, S.; Johnson, P.R.; MacDonald, P.E.; Gloyn, A.L.; Rorsman, P.; Braun, M. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic beta- and alpha-cells. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1107–E1116. [Google Scholar] [CrossRef] [Green Version]
- Gromada, J.; Hoy, M.; Buschard, K.; Salehi, A.; Rorsman, P. Somatostatin inhibits exocytosis in rat pancreatic alpha-cells by G(i2)-dependent activation of calcineurin and depriming of secretory granules. J. Physiol. 2001, 535, 519–532. [Google Scholar] [CrossRef]
- Hsu, W.H.; Xiang, H.D.; Rajan, A.S.; Kunze, D.L.; Boyd, A.E., 3rd. Somatostatin inhibits insulin secretion by a G-protein-mediated decrease in Ca2+ entry through voltage-dependent Ca2+ channels in the beta cell. J. Biol. Chem. 1991, 266, 837–843. [Google Scholar] [CrossRef]
- Samson, W.K.; Zhang, J.V.; Avsian-Kretchmer, O.; Cui, K.; Yosten, G.L.; Klein, C.; Lyu, R.M.; Wang, Y.X.; Chen, X.Q.; Yang, J.; et al. Neuronostatin encoded by the somatostatin gene regulates neuronal, cardiovascular, and metabolic functions. J. Biol. Chem. 2008, 283, 31949–31959. [Google Scholar] [CrossRef] [Green Version]
- Yosten, G.L.; Elrick, M.M.; Salvatori, A.; Stein, L.M.; Kolar, G.R.; Ren, J.; Corbett, J.A.; Samson, W.K. Understanding peptide biology: The discovery and characterization of the novel hormone, neuronostatin. Peptides 2015, 72, 192–195. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, A.S.; Elrick, M.M.; Samson, W.K.; Corbett, J.A.; Yosten, G.L. Neuronostatin inhibits glucose-stimulated insulin secretion via direct action on the pancreatic alpha-cell. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1257–E1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrick, M.M.; Samson, W.K.; Corbett, J.A.; Salvatori, A.S.; Stein, L.M.; Kolar, G.R.; Naatz, A.; Yosten, G.L. Neuronostatin acts via GPR107 to increase cAMP-independent PKA phosphorylation and proglucagon mRNA accumulation in pancreatic alpha-cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R143–R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broglio, F.; Arvat, E.; Benso, A.; Gottero, C.; Prodam, F.; Grottoli, S.; Papotti, M.; Muccioli, G.; van der Lely, A.J.; Deghenghi, R.; et al. Endocrine activities of cortistatin-14 and its interaction with GHRH and ghrelin in humans. J. Clin. Endocrinol. Metab. 2002, 87, 3783–3790. [Google Scholar] [CrossRef]
- Gahete, M.D.; Duran-Prado, M.; Luque, R.M.; Martinez-Fuentes, A.J.; Vazquez-Martinez, R.; Malagon, M.M.; Castano, J.P. Are somatostatin and cortistatin two siblings in regulating endocrine secretions? In vitro work ahead. Mol. Cell. Endocrinol. 2008, 286, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Castellano-Munoz, M.; Rafacho, A.; Alonso-Magdalena, P.; Marroqui, L.; Ruiz-Pino, A.; Bru-Tari, E.; Merino, B.; Irles, E.; Bello-Perez, M.; et al. Cortistatin regulates glucose-induced electrical activity and insulin secretion in mouse pancreatic beta-cells. Mol. Cell. Endocrinol. 2019, 479, 123–132. [Google Scholar] [CrossRef]
- Allia, E.; Tarabra, E.; Volante, M.; Cerrato, M.; Ghigo, E.; Muccioli, G.; Papotti, M. Expression of cortistatin and MrgX2, a specific cortistatin receptor, in human neuroendocrine tissues and related tumours. J. Pathol. 2005, 207, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Dorrell, C.; Schug, J.; Lin, C.F.; Canaday, P.S.; Fox, A.J.; Smirnova, O.; Bonnah, R.; Streeter, P.R.; Stoeckert, C.J., Jr.; Kaestner, K.H.; et al. Transcriptomes of the major human pancreatic cell types. Diabetologia 2011, 54, 2832–2844. [Google Scholar] [CrossRef] [Green Version]
- Papotti, M.; Tarabra, E.; Allia, E.; Bozzalla-Cassione, F.; Broglio, F.; Deghenghi, R.; Ghigo, E.; Muccioli, G. Presence of cortistatin in the human pancreas. J. Endocrinol. Investig. 2003, 26, RC15–RC18. [Google Scholar] [CrossRef]
- Unger, R.H. Glucagon and insulin: A bihormonal system. Compr. Ther. 1976, 2, 20–26. [Google Scholar]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portela-Gomes, G.M.; Grimelius, L.; Westermark, P.; Stridsberg, M. Somatostatin receptor subtypes in human type 2 diabetic islets. Pancreas 2010, 39, 836–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiter, E.H.; Gapp, D.A.; Eppig, J.J.; Coleman, D.L. Ultrastructural and morphometric studies of delta cells in pancreatic islets from C57BL/Ks diabetes mice. Diabetologia 1979, 17, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folli, F.; La Rosa, S.; Finzi, G.; Davalli, A.M.; Galli, A.; Dick, E.J., Jr.; Perego, C.; Mendoza, R.G. Pancreatic islet of Langerhans’ cytoarchitecture and ultrastructure in normal glucose tolerance and in type 2 diabetes mellitus. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 137–144. [Google Scholar] [CrossRef]
- Guardado Mendoza, R.; Perego, C.; Finzi, G.; La Rosa, S.; Capella, C.; Jimenez-Ceja, L.M.; Velloso, L.A.; Saad, M.J.; Sessa, F.; Bertuzzi, F.; et al. Delta cell death in the islet of Langerhans and the progression from normal glucose tolerance to type 2 diabetes in non-human primates (baboon, Papio hamadryas). Diabetologia 2015, 58, 1814–1826. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Halim, S.M.; Guenifi, A.; Efendic, S.; Ostenson, C.G. Both somatostatin and insulin responses to glucose are impaired in the perfused pancreas of the spontaneously noninsulin-dependent diabetic GK (Goto-Kakizaki) rats. Acta Physiol. Scand. 1993, 148, 219–226. [Google Scholar] [CrossRef]
- Weir, G.C.; Clore, E.T.; Zmachinski, C.J.; Bonner-Weir, S. Islet secretion in a new experimental model for non-insulin-dependent diabetes. Diabetes 1981, 30, 590–595. [Google Scholar] [CrossRef]
- Saisho, Y. beta-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 109–124. [Google Scholar] [CrossRef]
- Yue, J.T.; Riddell, M.C.; Burdett, E.; Coy, D.H.; Efendic, S.; Vranic, M. Amelioration of hypoglycemia via somatostatin receptor type 2 antagonism in recurrently hypoglycemic diabetic rats. Diabetes 2013, 62, 2215–2222. [Google Scholar] [CrossRef] [Green Version]
- Kellard, J.A.; Rorsman, N.J.G.; Hill, T.G.; Armour, S.L.; van de Bunt, M.; Rorsman, P.; Knudsen, J.G.; Briant, L.J.B. Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high-fat diet. Mol. Metab. 2020, 40, 101021. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, R.; Yang, T.; Zhang, Q. δ-Cells: The Neighborhood Watch in the Islet Community. Biology 2021, 10, 74. https://doi.org/10.3390/biology10020074
Gao R, Yang T, Zhang Q. δ-Cells: The Neighborhood Watch in the Islet Community. Biology. 2021; 10(2):74. https://doi.org/10.3390/biology10020074
Chicago/Turabian StyleGao, Rui, Tao Yang, and Quan Zhang. 2021. "δ-Cells: The Neighborhood Watch in the Islet Community" Biology 10, no. 2: 74. https://doi.org/10.3390/biology10020074
APA StyleGao, R., Yang, T., & Zhang, Q. (2021). δ-Cells: The Neighborhood Watch in the Islet Community. Biology, 10(2), 74. https://doi.org/10.3390/biology10020074