
Oxidative Pentose Phosphate Pathway Enzyme 6-Phosphogluconate Dehydrogenase Plays a Key Role in Breast Cancer Metabolism

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. siRNA Transfection
2.3. RNA Isolation and Gene Expression Analysis
2.4. Enzyme Activity Assays
2.4.1. 6-Phosphogluconate Dehydrogenase (6PGD), Malic Enzyme (ME) and Isocitrate Dehydrogenase (IDH)
2.4.2. Lactate Dehydrogenase
2.4.3. Transketolase
2.5. Cell Proliferation and Cell Cycle Distribution Analysis
2.6. Mammosphere Formation Assay (3D Cell Culture)
2.7. Western Blot
2.8. Metabolite Production and Consumption Analysis
2.9. Intracellular ROS Level Measurement
2.10. Apoptosis Measurement
2.11. Senescence-Associated-β Galactosidase Activity
2.12. Statistical Analysis
3. Results
3.1. 6PGD Was Successfully Inhibited in MCF7 Cells
3.2. 6PGD Knockdown Affects Cell Proliferation, Cell Cycle Distribution, and Apoptosis
3.3. p53 Is Upregulated with 6PGD Knockdown
3.4. NADPH Produced by 6PGD Is Dispensable for ROS Detoxification
3.5. 6PGD Inhibition Leads Breast Cancer Cells to Reprogram Their Central Carbon Metabolism
3.6. 6PGD Knockdown Reduces the Mammosphere Formation Capacity of Breast Cancer Cells
3.7. S3 Is a Specific 6PGD Inhibitor that Induces Senescence in MCF7 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xu, Y.; Chen, T.; Du, Z.; Liu, X.; Hu, Z.; Wei, D.; Gao, C.; Zhang, W.; Li, Q. Targeting oxidative pentose phosphate pathway prevents recurrence in mutant Kras colorectal carcinomas. PLoS Biol. 2019, 17, e3000425. [Google Scholar] [CrossRef] [PubMed]
- Vizán, P.; Alcarraz-Vizán, G.; Díaz-Moralli, S.; Solovjeva, O.N.; Frederiks, W.M.; Cascante, M. Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29. Int. J. Cancer 2009, 124, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Sukhatme, V.P.; Chan, B. Glycolytic cancer cells lacking 6-phosphogluconate dehydro-genase metabolize glucose to induce senescence. FEBS Lett. 2012, 586, 2389–2395. [Google Scholar] [CrossRef] [Green Version]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free. Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors (Review). Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Montoya, A.; Lee, W.-N.P.; Bassilian, S.; Lim, S.; Trebukhina, R.V.; Kazhyna, M.V.; Ciudad, C.J.; Noé, V.; Centelles, J.J.; Cascante, M. Pentose phosphate cycle oxidative and nonoxidative balance: A new vulnerable target for overcoming drug resistance in cancer. Int. J. Cancer 2006, 119, 2733–2741. [Google Scholar] [CrossRef] [PubMed]
- De Preter, G.; Neveu, M.-A.; Danhier, P.; Brisson, L.; Payen, V.L.; Porporato, P.E.; Jordan, B.F.; Sonveaux, P.; Gallez, B. Inhibition of the pentose phosphate pathway by dichloroacetate un-ravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget 2015, 19, 2910–2920. [Google Scholar]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Re-sistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar]
- Jia, P.; Dai, C.; Cao, P.; Sun, D.; Ouyang, R.; Miao, Y. The role of reactive oxygen species in tumor treatment. RSC Adv. 2020, 10, 7740–7750. [Google Scholar] [CrossRef]
- Hartmannsberger, D.; Mack, B.; Eggert, C.; Denzel, S.; Stepp, H.; Betz, C.S.; Gires, O. Transketolase-like protein 1 confers resistance to serum with-drawal in vitro. Cancer Lett. 2011, 300, 20–29. [Google Scholar]
- Jonas, S.K.; Benedetto, C.; Flatman, A.; Hammond, R.H.; Micheletti, L.; Riley, C.; Riley, P.A.; Spargo, D.J.; Zonca, M.; Slater, T.F. Increased activity of 6-phosphogluconate dehydrogenase and glu-cose-6-phosphate dehydrogenase in purified cell suspensions and single cells from the uterine cervix in cervical intraepithelial neoplasia. Br. J. Cancer 1992, 66, 185–191. [Google Scholar]
- Lin, C.-J.; Ho, H.-Y.; Cheng, M.-L.; Cheng, T.-H.; Yu, J.-S.; Chiu, D.T.-Y. Impaired dephosphorylation renders G6PD-knockdown HepG2 cells more susceptible to H2O2-induced apoptosis. Free. Radic. Biol. Med. 2010, 49, 361–373. [Google Scholar] [CrossRef]
- Benito, A.; Polat, I.H.; Noé, V.; Ciudad, C.J.; Marin, S.; Cascante, M. Glucose-6-phosphate dehydrogenase and transketolase modulate breast cancer cell metabolic reprogramming and correlate with poor patient outcome. Oncotarget 2017, 8, 106693–106706. [Google Scholar] [CrossRef]
- Lin, R.; Elf, S.; Shan, C.; Kang, H.-B.; Ji, Q.; Zhou, L.; Hitosugi, T.; Zhang, L.; Zhang, S.; Seo, J.H.; et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat. Cell Biol. 2015, 17, 1484–1496. [Google Scholar] [CrossRef] [Green Version]
- Sarfraz, I.; Rasul, A.; Hussain, G.; Shah, M.A.; Zahoor, A.F.; Asrar, M.; Selamoglu, Z.; Ji, X.; Adem, Ş.; Sarker, S.D. 6-Phosphogluconate dehydrogenase fuels multiple aspects of cancer cells: From cancer initiation to metastasis and chemoresistance. BioFactors 2020, 46, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Peng, X.; Huang, J. Inhibiting 6-phosphogluconate dehydrogenase selectively targets breast cancer through AMPK activation. Clin. Transl. Oncol. 2018, 20, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, C.; Elf, S.; Ji, Q.; Kang, H.-B.; Zhou, L.; Hitosugi, T.; Jin, L.; Lin, R.; Zhang, L.; Seo, J.H.; et al. Lysine Acetylation Activates 6-Phosphogluconate Dehydrogenase to Promote Tumor Growth. Mol. Cell 2014, 55, 552–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Zhao, L.; Liu, S.; Li, Y.; Xia, S.; Chen, D.; Wang, M.; Wu, S.; Dai, Q.; Vu, H.; et al. γ-6-Phosphogluconolactone, a Byproduct of the Oxidative Pentose Phosphate Pathway, Contributes to AMPK Activation through Inhibition of PP2A. Mol. Cell 2019, 76, 857–871.e9. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xiang, Z.; Zhang, Y.; Sun, D. Inhibiting 6-phosphogluconate dehydrogenase enhances chemotherapy ef-ficacy in cervical cancer via AMPK-independent inhibition of RhoA and Rac1. Clin. Transl. Oncol. 2019, 21, 404–411. [Google Scholar] [PubMed]
- Cao, J.; Sun, X.; Zhang, X.; Chen, D. 6PGD Upregulation is Associated with Chemo- and Immuno-Resistance of Renal Cell Carcinoma via AMPK Signaling-Dependent NADPH-Mediated Metabolic Repro-graming. Am. J. Med. Sci. 2020, 360, 279–286. [Google Scholar]
- Chen, H.; Wu, D.; Bao, L.; Yin, T.; Lei, D.; Yu, J.; Tong, X. 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed. Pharmacother. 2019, 111, 1353–1358. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glu-cose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, V.; He, J. Reactive Oxygen Species, Metabolic Plasticity, and Drug Resistance in Cancer. Int. J. Mol. Sci. 2020, 21, 3412. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Hammill, A.K.; Uhr, J.W.; Scheuermann, R.H. Annexin V staining due to loss of mem-brane asymmetry can be reversible and precede commitment to apoptotic death. Exp. Cell Res. 1999, 251, 16–21. [Google Scholar] [PubMed]
- Zanuy, M.; Ramos-Montoya, A.; Villacañas, O.; Canela, N.; Miranda, A.; Aguilar, E.; Agell, N.; Bachs, O.; Rubio-Martínez, J.; Pujol, M.D.; et al. Cyclin-dependent kinases 4 and 6 control tumor progression and direct glucose oxidation in the pentose cycle. Metabolomics 2011, 8, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013, 138, 255–271. [Google Scholar] [CrossRef]
- Pucci, B.; Kasten, M.; Giordano, A. Cell Cycle and Apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Bratton, D.L.; Dreyer, E.; Kailey, J.M.; Fadok, V.A.; Clay, K.L.; Henson, P.M. The mechanism of internalization of platelet-activating factor in acti-vated human neutrophils. Enhanced transbilayer movement across the plasma membrane. J. Immunol. 1992, 148, 514–523. [Google Scholar]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Vojtĕsek, B.; Lane, D.P. Regulation of p53 protein expression in human breast cancer cell lines. J. Cell Sci. 1993, 105, 607–612. [Google Scholar] [PubMed]
- Ahn, W.S.; Antoniewicz, M.R. Parallel labeling experiments with [1,2-13C]glucose and [U-13C]glutamine provide new insights into CHO cell metabolism. Metab. Eng. 2013, 15, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Zancan, P.; Sola-Penna, M.; Furtado, C.M.; Da Silva, D. Differential expression of phosphofructokinase-1 isoforms correlates with the glycolytic efficiency of breast cancer cells. Mol. Genet. Metab. 2010, 100, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Kung, H.-N.; Marks, J.R.; Chi, J.-T.A. Glutamine Synthetase Is a Genetic Determinant of Cell Type–Specific Glutamine Independence in Breast Epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, T. Stem cell-like cancer cells in cancer cell lines. Cancer Biomark. 2007, 3, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Jerby, L.; Wolf, L.; Denkert, C.; Stein, G.Y.; Hilvo, M.; Oresic, M.; Geiger, T.; Ruppin, E. Metabolic associations of reduced proliferation and oxidative stress in advanced breast cancer. Cancer Res. 2012, 72, 5712–5720. [Google Scholar] [CrossRef] [Green Version]
- Prieur, A.; Besnard, E.; Babled, A.; Lemaitre, J.-M. p53 and p16INK4A independent induction of senescence by chroma-tin-dependent alteration of S-phase progression. Nat. Commun. 2011, 2, 473. [Google Scholar] [CrossRef]
- Hong, S.M.; Park, C.W.; Kim, S.W.; Nam, Y.J.; Yu, J.H.; Shin, J.H.; Yun, C.H.; Im, S.-H.; Kim, K.-T.; Sung, Y.C.; et al. NAMPT suppresses glucose deprivation-induced oxidative stress by in-creasing NADPH levels in breast cancer. Oncogene 2015, 35, 3544–3554. [Google Scholar] [CrossRef]
- Blanquer-Rossello Mdel, M.; Olivera, J.; Sastre-Serra, J.; Valle, A.; Roca, P. Leptin regulates energy metabolism in MCF-7 breast cancer cells. Int. J. Biochem. Cell Biol. 2016, 72, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Li, X.; Sun, W.; Li, Y.; Yuan, Y.; Guan, B.; Zhang, S. Discovery of Ebselen as an Inhibitor of 6PGD for Suppressing Tumor Growth. Cancer Manag. Res. 2020, 12, 6921–6934. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Adler, J.; Meltser, V.; Reuven, N.; Shaul, Y. AMPK couples p73 with p53 in cell fate decision. Cell Death Differ. 2014, 21, 1451–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filosa, S.; Fico, A.; Paglialunga, F.; Balestrieri, M.; Crooke, A.; Verde, P.; Abrescia, P.; Bautista, J.M.; Martini, G. Failure to increase glucose consumption through the pentose-phosphate pathway results in the death of glucose-6-phosphate dehydrogenase gene-deleted mouse em-bryonic stem cells subjected to oxidative stress. Biochem. J. 2003, 370, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995, 14, 5209–5215. [Google Scholar] [CrossRef]
- Smith, S.B.; Freedland, R.A. Activation of pyruvate kinase by 6-phosphogluconate. J. Biol. Chem. 1979, 254, 10644–10648. [Google Scholar] [CrossRef]
- Sommercorn, J.; Freedland, R.A. Regulation of hepatic phosphofructokinase by 6-phosphogluconate. J. Biol. Chem. 1982, 257, 9424–9428. [Google Scholar] [CrossRef]
- Muller, F.L.; Aquilanti, E.A.; Depinho, R.A. Collateral Lethality: A New Therapeutic Strategy in Oncology. Trends Cancer 2015, 1, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Heiden, M.G.V.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef]
- Heiden, M.G.V. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Vikas, P.; Sukhatme, C.B.C. Methods and Compositions for 6-Phosphogluconate de-Hydrogenase (6-pgd) as a Target for Lung Cancer Therapy. U.S. Patent No. WO2013152186A8, 1 May 2014. [Google Scholar]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Baker, D.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.; VanderLaan, P.A.; Sukhatme, V.P. 6-Phosphogluconate dehydrogenase regu-lates tumor cell migration in vitro by regulating receptor tyrosine kinase c-Met. Biochem. Biophys. Res. Commun. 2013, 439, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer Cell Lines Are Useful Model Systems for Medical Research. Cancers 2019, 11, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
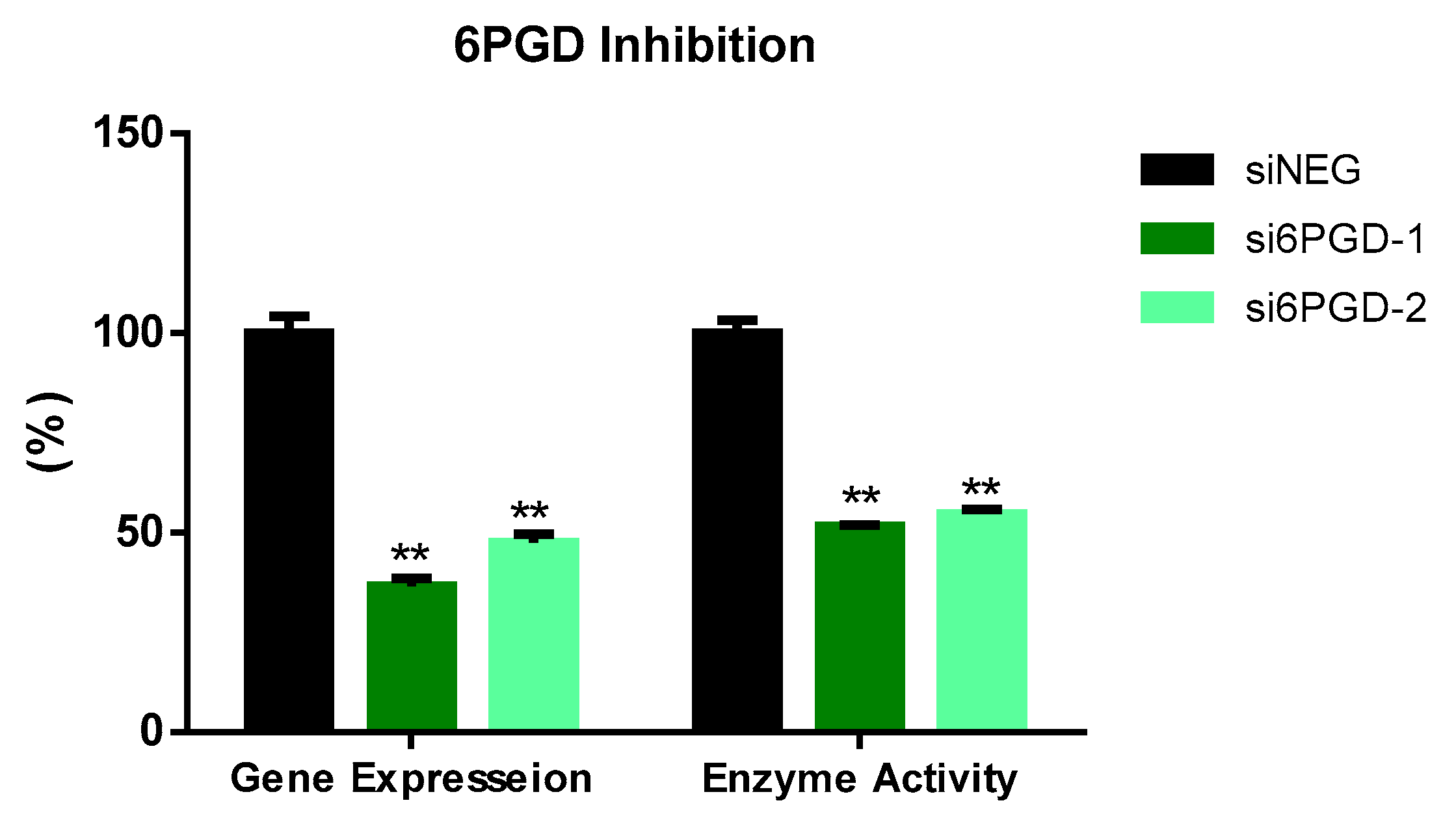




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polat, I.H.; Tarrado-Castellarnau, M.; Bharat, R.; Perarnau, J.; Benito, A.; Cortés, R.; Sabatier, P.; Cascante, M. Oxidative Pentose Phosphate Pathway Enzyme 6-Phosphogluconate Dehydrogenase Plays a Key Role in Breast Cancer Metabolism. Biology 2021, 10, 85. https://doi.org/10.3390/biology10020085
Polat IH, Tarrado-Castellarnau M, Bharat R, Perarnau J, Benito A, Cortés R, Sabatier P, Cascante M. Oxidative Pentose Phosphate Pathway Enzyme 6-Phosphogluconate Dehydrogenase Plays a Key Role in Breast Cancer Metabolism. Biology. 2021; 10(2):85. https://doi.org/10.3390/biology10020085
Chicago/Turabian StylePolat, Ibrahim H., Míriam Tarrado-Castellarnau, Rohit Bharat, Jordi Perarnau, Adrian Benito, Roldán Cortés, Philippe Sabatier, and Marta Cascante. 2021. "Oxidative Pentose Phosphate Pathway Enzyme 6-Phosphogluconate Dehydrogenase Plays a Key Role in Breast Cancer Metabolism" Biology 10, no. 2: 85. https://doi.org/10.3390/biology10020085
APA StylePolat, I. H., Tarrado-Castellarnau, M., Bharat, R., Perarnau, J., Benito, A., Cortés, R., Sabatier, P., & Cascante, M. (2021). Oxidative Pentose Phosphate Pathway Enzyme 6-Phosphogluconate Dehydrogenase Plays a Key Role in Breast Cancer Metabolism. Biology, 10(2), 85. https://doi.org/10.3390/biology10020085