One Year of SARS-CoV-2: How Much Has the Virus Changed?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Sequence Data and Residue Mutation Rates
2.2. Temporal Analysis
2.3. Temporal/Geographical Analysis
2.4. Protein Structure-Based Mutational Analysis
3. Results and Discussion
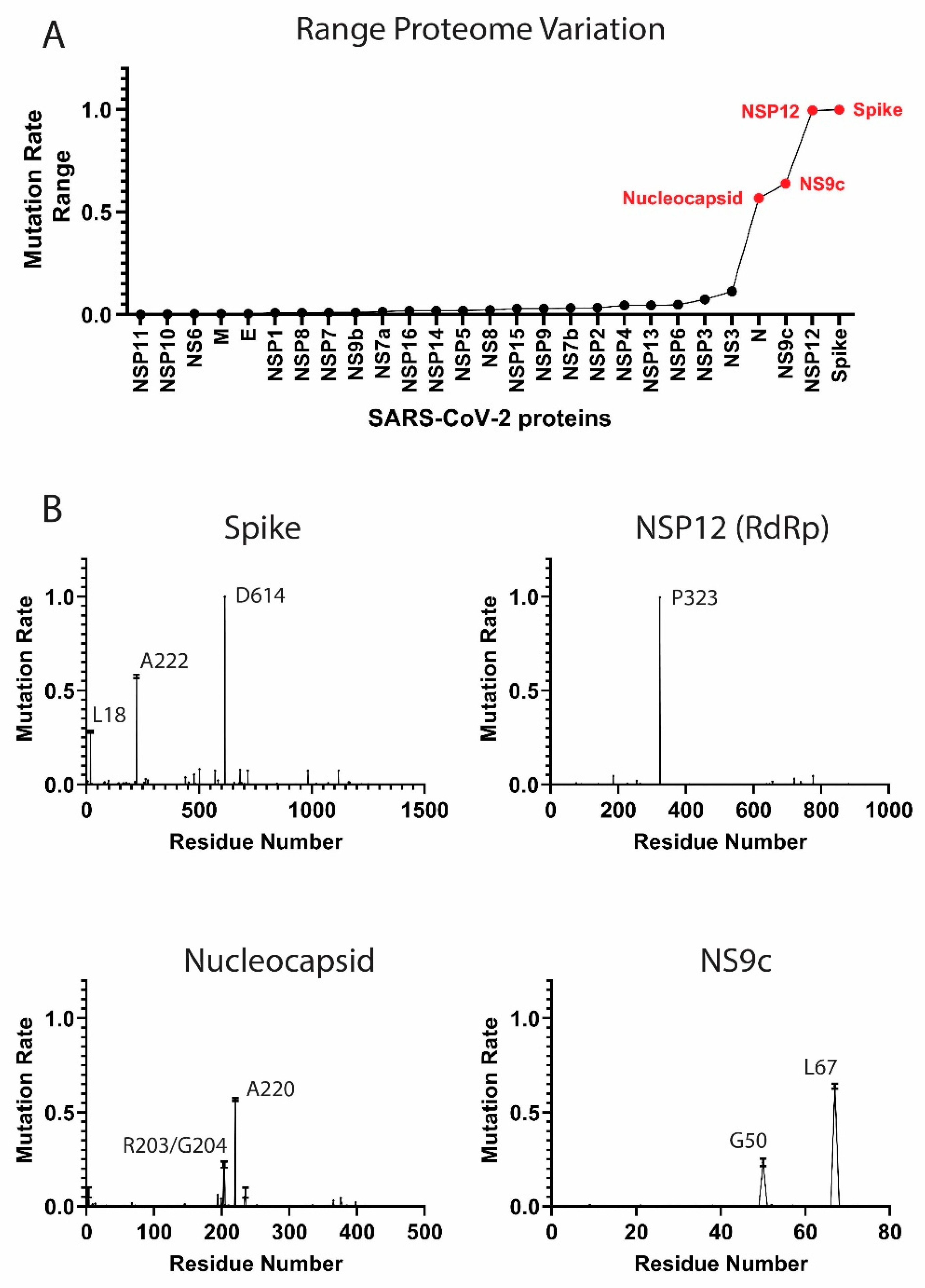
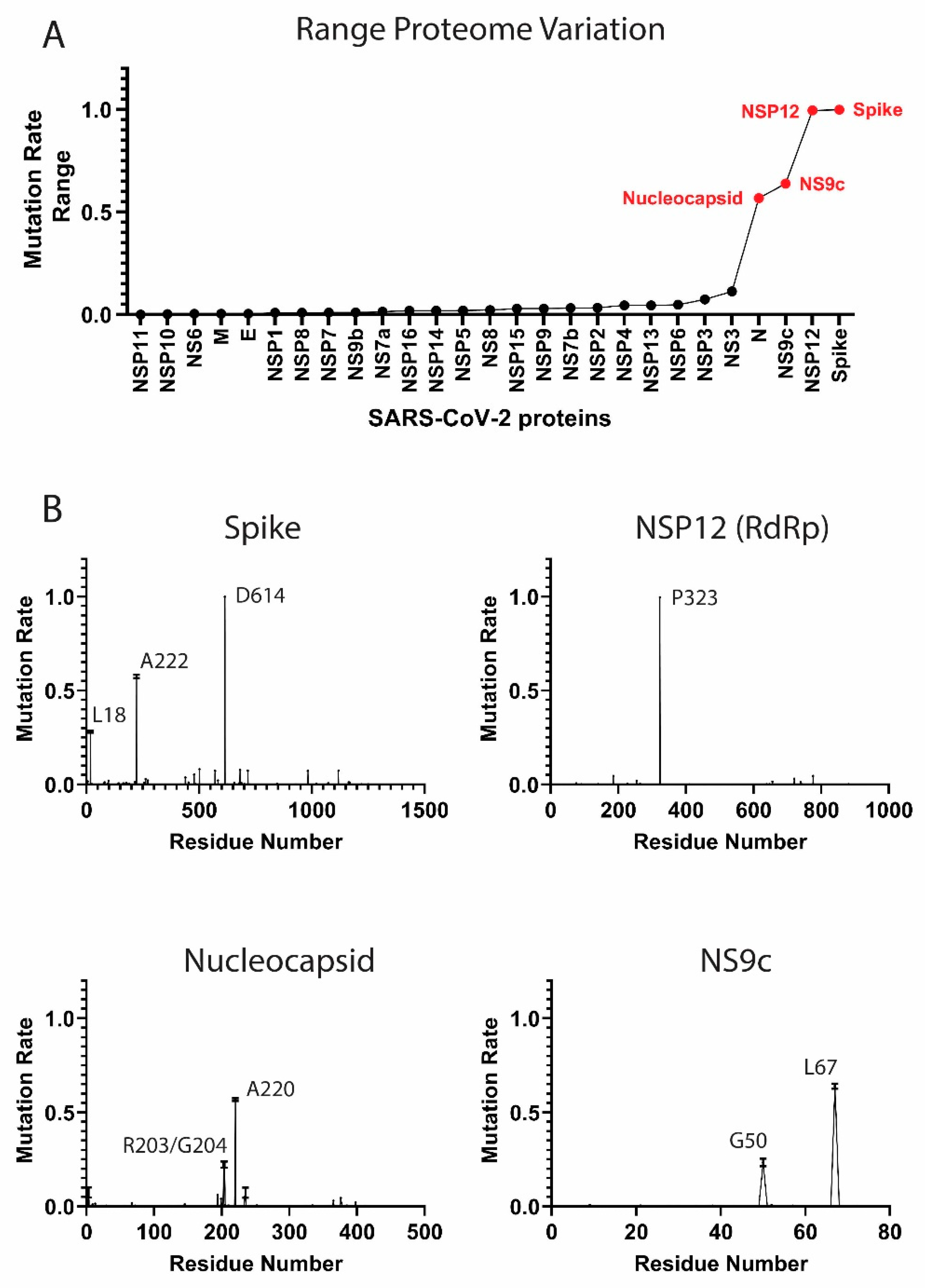
3.1. Components of the SARS-CoV-2 Proteome Are Mutating at Different Rates
High-Frequency Mutating SARS-CoV-2 Proteome Components
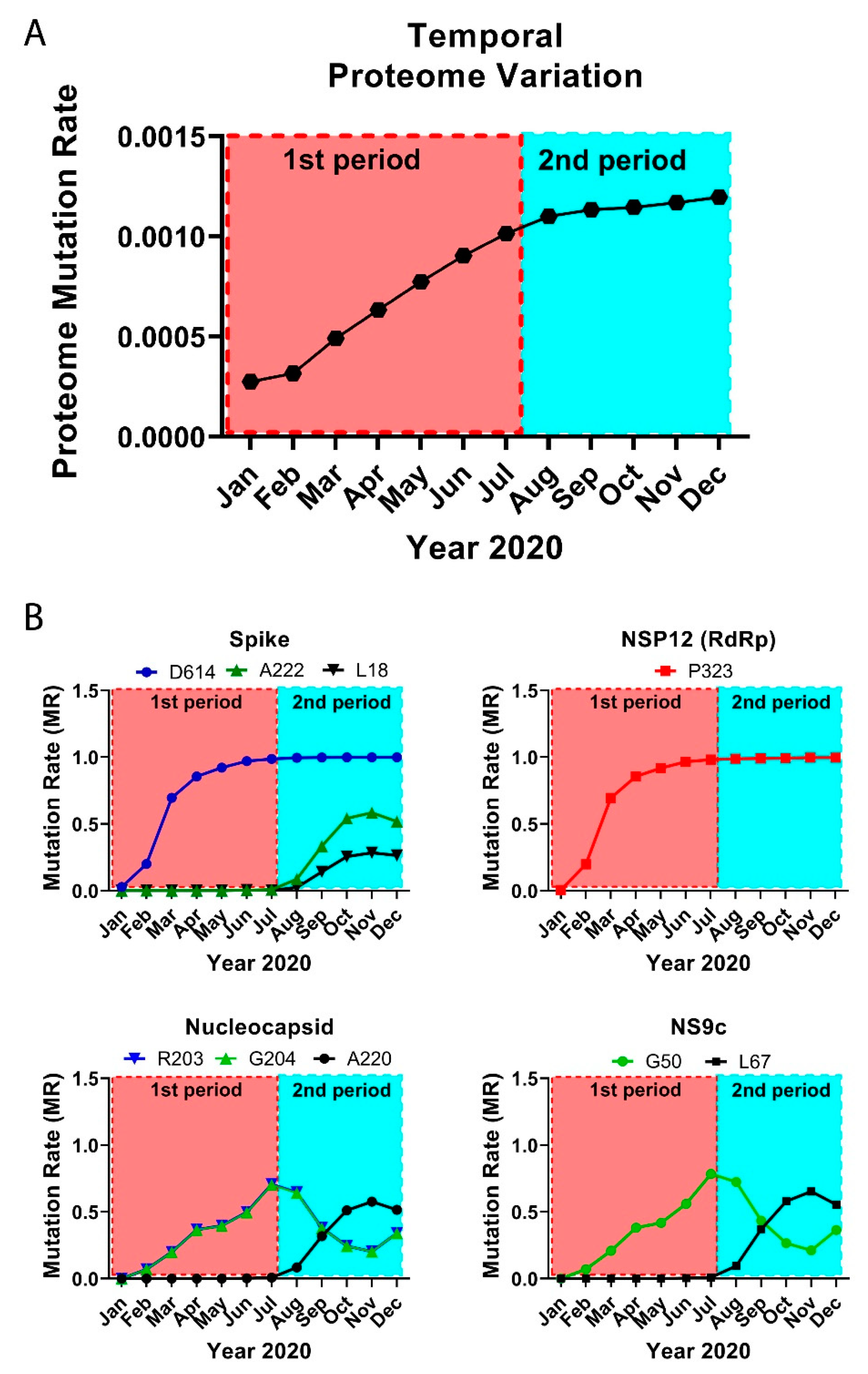
3.2. The Temporal Emergence of Proteome Mutations
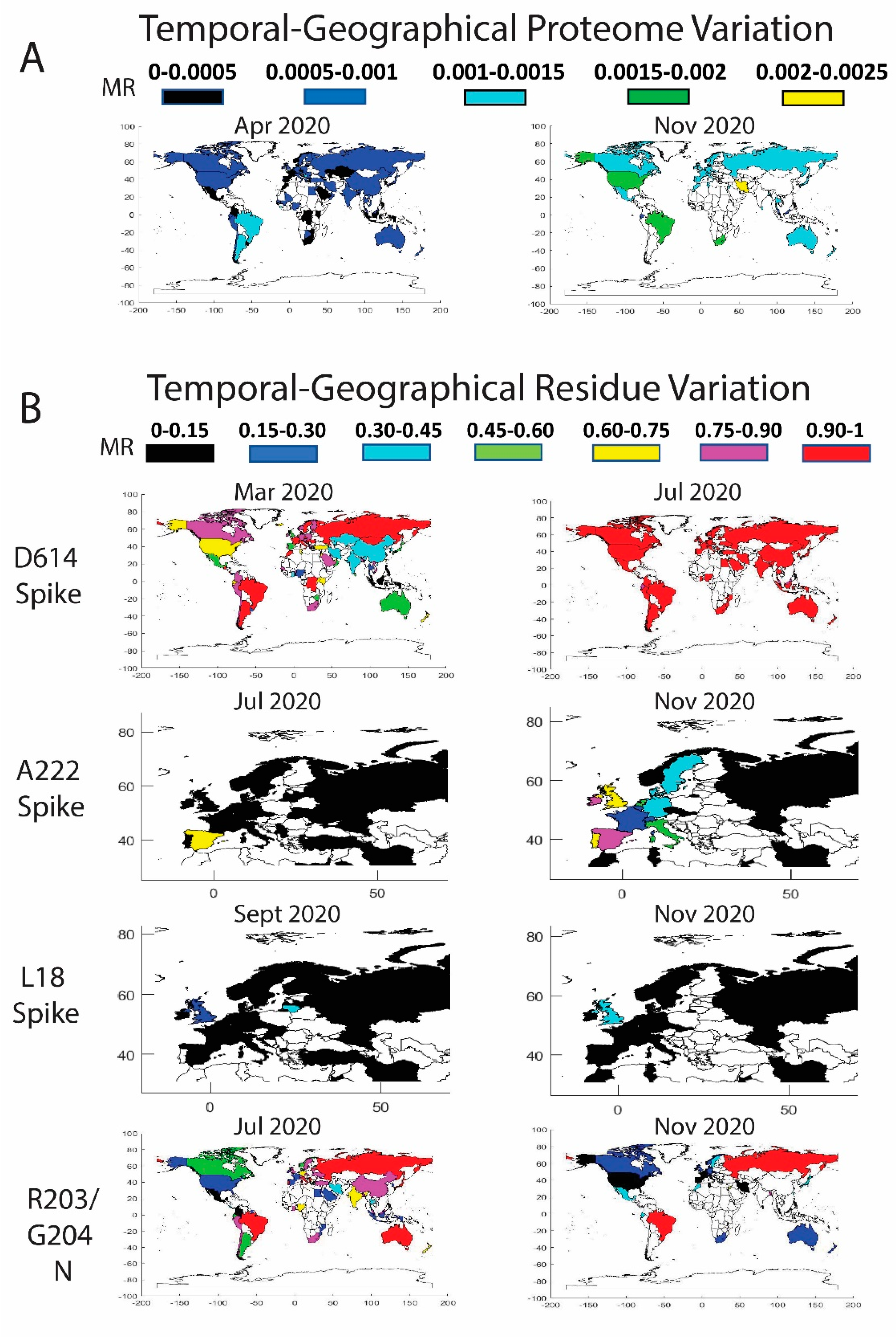
3.3. Worldwide Geographical and Temporal Differences in Proteome Variation
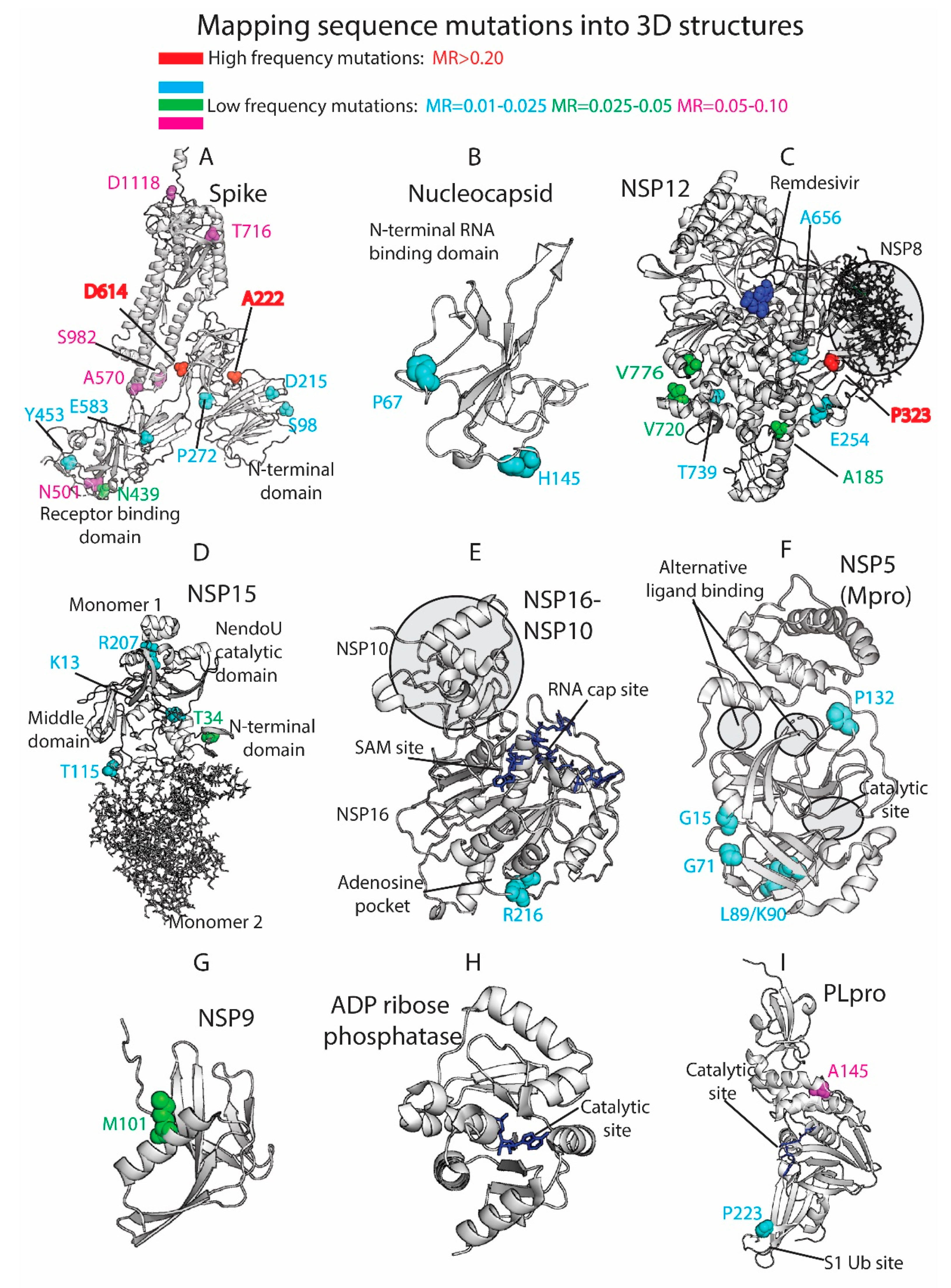
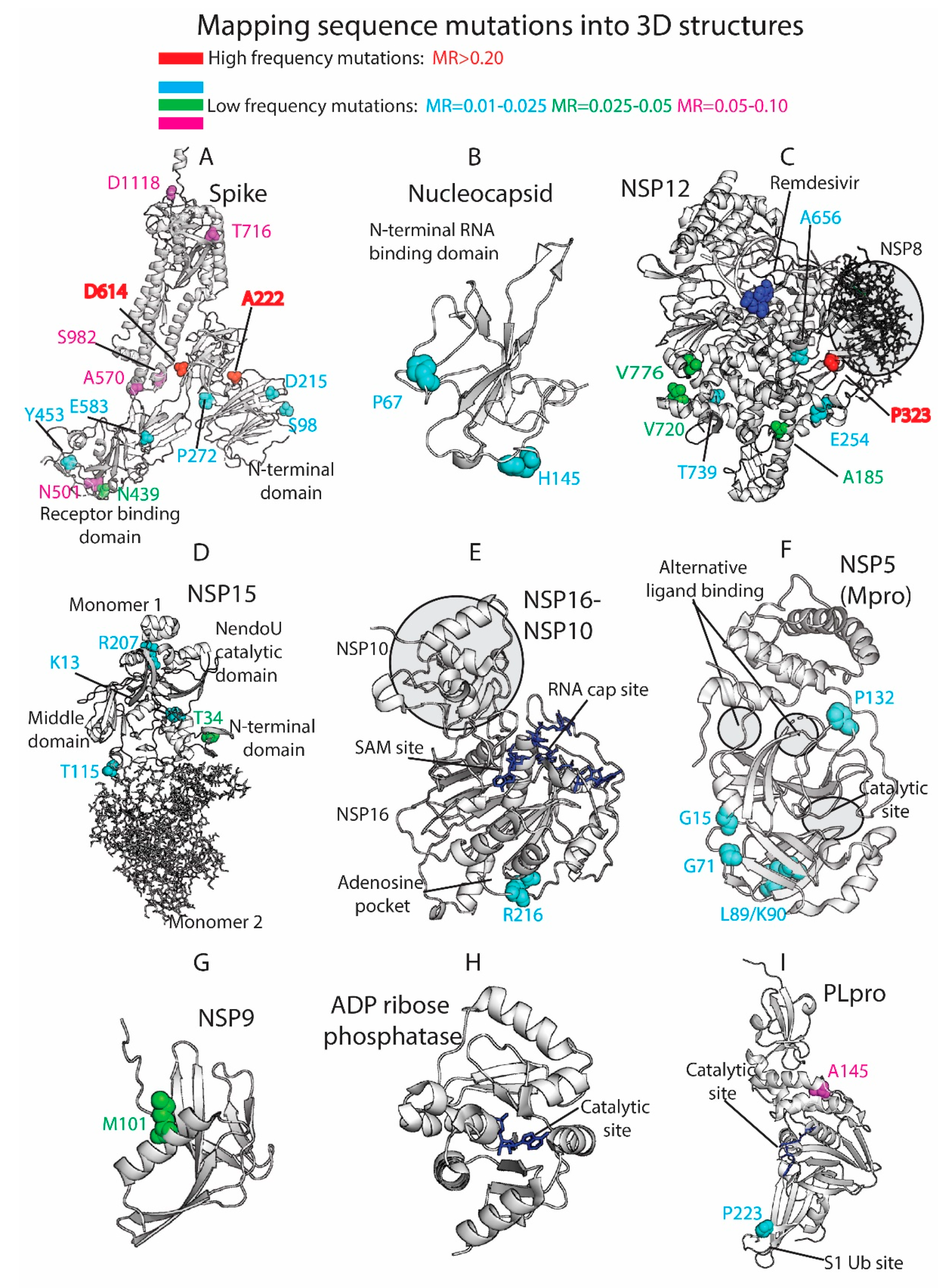
3.4. Residue Variation at 3D Molecular Level: Mapping into Crystallized Proteins
3.5. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MR | Mutation rate |
| S | Spike |
| N | Nucleocapsid |
| E | Envelope |
| M | Membrane |
| RdRp | RNA-dependent RNA polymerase |
| PLpro | Papain-like protease |
| Mpro | Main protease |
References
- Doherty, P.C. What have we learnt so far from COVID-19? Nat. Rev. Immunol. 2021, 1–2. [Google Scholar] [CrossRef]
- Dow, A.W.; DiPiro, J.T.; Giddens, J.; Buckley, P.; Santen, S.A. Emerging From the COVID-19 Crisis With a Stronger Health Care Workforce. Acad. Med. 2020, 95, 1823–1826. [Google Scholar] [CrossRef]
- OECD Policy Responses to Coronavirus (COVID-19). The Territorial Impact of COVID-19: Managing the Crisis Across Levels of Government. Available online: http://www.oecd.org/coronavirus/policy-responses/the-territorial-impact-of-covid-19-managing-the-crisis-across-levels-of-government-d3e314e1/#section-d1e343 (accessed on 15 December 2020).
- Morawska, L.; Cao, J. Airborne transmission of SARS-CoV-2: The world should face the reality. Environ. Int. 2020, 139, 105730. [Google Scholar] [CrossRef] [PubMed]
- Race, M.; Ferraro, A.; Galdiero, E.; Guida, M.; Núñez-Delgado, A.; Pirozzi, F.; Siciliano, A.; Fabbricino, M. Current emerging SARS-CoV-2 pandemic: Potential direct/indirect negative impacts of virus persistence and related therapeutic drugs on the aquatic compartments. Environ. Res. 2020, 188, 109808. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Allergy and Infectious Diseases (NIAID). COVID-19 Is an Emerging, Rapidly Evolving Situation. Available online: https://www.niaid.nih.gov/diseases-conditions/coronaviruses (accessed on 2 September 2020).
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.-I.; Hsueh, P.-R. Emerging threats from zoonotic coronaviruses-from SARS and MERS to 2019-nCoV. J. Microbiol. Immunol. Infect. 2020, 53, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comandatore, F.; Chiodi, A.; Gabrieli, P.; Biffignandi, G.B.; Perini, M.; Ramazzotti, M.; Ricagno, S.; Rimoldi, S.G.; Gismondo, M.R.; Micheli, V.; et al. Identification of variable sites in Sars-CoV-2 and their abundance profiles in time. bioRxiv 2020. [Google Scholar] [CrossRef]
- Khan, M.I.; Khan, Z.A.; Baig, M.H.; Ahmad, I.; Farouk, A.E.; Song, Y.G.; Dong, J.J. Comparative genome analysis of novel coronavirus (SARS-CoV-2) from different geographical locations and the effect of mutations on major target proteins: An in silico insight. PLoS ONE 2020, 15, e0238344. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Patro, P.P.; Sathyaseelan, C.; Uttamrao, P.P.; Rathinavelan, T. Global variation in the SARS-CoV-2 proteome reveals the mutational hotspots in the drug and vaccine candidates. bioRxiv 2020. [Google Scholar] [CrossRef]
- GISAID, the Global Initiative on Sharing All Influenza Data. Available online: https://www.gisaid.org/ (accessed on 2 November 2020).
- RCSB Protein Data Bank. Available online: http://www.rcsb.org/ (accessed on 2 September 2020).
- Python Software Foundation. Python Language Reference. Available online: http://www.python.org (accessed on 2 July 2020).
- MathWorks, Inc. MATLAB: The Language of Technical Computing: Computation, Visualization, Programming. 1996. Available online: https://www.mathworks.com/ (accessed on 2 August 2020).
- Our World in Data. Statistics and Research. Coronavirus (COVID-19) Cases. Available online: https://github.com/owid/covid-19-data/tree/master/public/data (accessed on 2 September 2020).
- The PyMOL Molecular Graphics System, Version 2.4.0; Schrödinger, LLC.: New York, NY, USA, 2010.
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- ECDC. Rapid Increase of a SARS-CoV-2 Variant with Multiple Spike Protein Mutations Observed in the United Kingdom European Centre for Disease Prevention and Control; ECDC: Solna kommun, Sweden, 2020. [Google Scholar]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Hsin, W.-C.; Chang, C.-H.; Chang, C.-Y.; Peng, W.-H.; Chien, C.-L.; Chang, M.-F.; Chang, S.C. Nucleocapsid protein-dependent assembly of the RNA packaging signal of Middle East respiratory syndrome coronavirus. J. Biomed. Sci. 2018, 25, 47. [Google Scholar] [CrossRef]
- Ayub, M.I. Reporting Two SARS-CoV-2 Strains Based on A Unique Trinucleotide-Bloc Mutation and Their Potential Pathogenic Difference. Preprints 2020. [Google Scholar] [CrossRef] [Green Version]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Comas, I.; González Candelas, F.; Stadler, T.; Neher, R.A. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv 2020. [Google Scholar] [CrossRef]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Non-Synonymous Mutations and Polyproline Regions. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Merow, C.; Urban, M.C. Seasonality and uncertainty in global COVID-19 growth rates. Proc. Natl. Acad. Sci. USA 2020, 117, 27456–27464. [Google Scholar] [CrossRef]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pract. 2020, 74, e13525. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Rosenberg, S.M. Reverse Mutation. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 220–221. [Google Scholar]
- Tortorici, M.A.; Veesler, D. Chapter Four—Structural insights into coronavirus entry. In Advances in Virus Research; Rey, F.A., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 105, pp. 93–116. [Google Scholar]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Mukherjee, R. Global efforts on vaccines for COVID-19: Since, sooner or later, we all will catch the coronavirus. J. Biosci. 2020, 45, 68. [Google Scholar] [CrossRef]
- Rowe, J.B.; Kapolka, N.J.; Taghon, G.J.; Morgan, W.M.; Isom, D.G. The evolution and mechanism of GPCR proton sensing. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Isom, D.G.; Dohlman, H.G. Buried ionizable networks are an ancient hallmark of G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA 2015, 112, 5702–5707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isom, D.G.; Sridharan, V.; Dohlman, H.G. Regulation of Ras Paralog Thermostability by Networks of Buried Ionizable Groups. Biochemistry 2016, 55, 534–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isom, D.G.; Sridharan, V.; Baker, R.; Clement, S.T.; Smalley, D.M.; Dohlman, H.G. Protons as second messenger regulators of G protein signaling. Mol. Cell 2013, 51, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Helenius, A. Virus entry: What has pH got to do with it? Nat. Cell Biol. 2013, 15, 125. [Google Scholar] [CrossRef]
- Zhang, B.-Z.; Hu, Y.-F.; Chen, L.-L.; Yau, T.; Tong, Y.-G.; Hu, J.-C.; Cai, J.-P.; Chan, K.-H.; Dou, Y.; Deng, J.; et al. Mining of epitopes on spike protein of SARS-CoV-2 from COVID-19 patients. Cell Res. 2020, 30, 702–704. [Google Scholar] [CrossRef]
- Liu, M.; Gu, C.; Wu, J.; Zhu, Y. Amino acids 1 to 422 of the spike protein of SARS associated coronavirus are required for induction of cyclooxygenase-2. Virus Genes 2006, 33, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein. Materials 2020, 13, 5362. [Google Scholar] [CrossRef]
- Moreira, R.A.; Chwastyk, M.; Baker, J.L.; Guzman, H.V.; Poma, A.B. Quantitative determination of mechanical stability in the novel coronavirus spike protein. Nanoscale 2020, 12, 16409–16413. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J.; et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldhill, D.H.; Te Velthuis, A.J.W.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delang, L.; Froeyen, M.; Herdewijn, P.; Neyts, J. Identification of a novel resistance mutation for benzimidazole inhibitors of the HCV RNA-dependent RNA polymerase. Antivir. Res. 2012, 93, 30–38. [Google Scholar] [CrossRef]
- Chand, G.B.; Banerjee, A.; Azad, G.K. Identification of novel mutations in RNA-dependent RNA polymerases of SARS-CoV-2 and their implications on its protein structure. PeerJ 2020, 8, e9492. [Google Scholar] [CrossRef] [PubMed]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [Green Version]
- Guarino, L.A.; Bhardwaj, K.; Dong, W.; Sun, J.; Holzenburg, A.; Kao, C. Mutational analysis of the SARS virus Nsp15 endoribonuclease: Identification of residues affecting hexamer formation. J. Mol. Biol. 2005, 353, 1106–1117. [Google Scholar] [CrossRef]
- Viswanathan, T.; Arya, S.; Chan, S.-H.; Qi, S.; Dai, N.; Misra, A.; Park, J.-G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef]
- Almazán, F.; Dediego, M.L.; Galán, C.; Escors, D.; Alvarez, E.; Ortego, J.; Sola, I.; Zuñiga, S.; Alonso, S.; Moreno, J.L.; et al. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. J. Virol. 2006, 80, 10900–10906. [Google Scholar] [CrossRef] [Green Version]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genomics 2020, 47, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Sutton, G.; Fry, E.; Carter, L.; Sainsbury, S.; Walter, T.; Nettleship, J.; Berrow, N.; Owens, R.; Gilbert, R.; Davidson, A.; et al. The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure 2004, 12, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Michalska, K.; Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Stols, L.; Endres, M.; Joachimiak, A. Crystal structures of SARS-CoV-2 ADP-ribose phosphatase: From the apo form to ligand complexes. bioRxiv 2020, 7, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. bioRxiv 2020. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The Papain-Like Protease of Severe Acute Respiratory Syndrome Coronavirus Has Deubiquitinating Activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [Green Version]
- SoRelle, J.A.; Frame, I.; Falcon, A.; Jacob, J.; Wagenfuehr, J.; Mitui, M.; Park, J.Y.; Filkins, L. Clinical Validation of a SARS-CoV-2 Real-Time Reverse Transcription PCR Assay Targeting the Nucleocapsid Gene. J. Appl. Lab. Med. 2020, 5, 889–896. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilar, S.; Isom, D.G. One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology 2021, 10, 91. https://doi.org/10.3390/biology10020091
Vilar S, Isom DG. One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology. 2021; 10(2):91. https://doi.org/10.3390/biology10020091
Chicago/Turabian StyleVilar, Santiago, and Daniel G. Isom. 2021. "One Year of SARS-CoV-2: How Much Has the Virus Changed?" Biology 10, no. 2: 91. https://doi.org/10.3390/biology10020091
APA StyleVilar, S., & Isom, D. G. (2021). One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology, 10(2), 91. https://doi.org/10.3390/biology10020091