Targeted Double-Stranded cDNA Sequencing-Based Phase Analysis to Identify Compound Heterozygous Mutations and Differential Allelic Expression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient and Sample
2.2. Genomic DNA Extraction
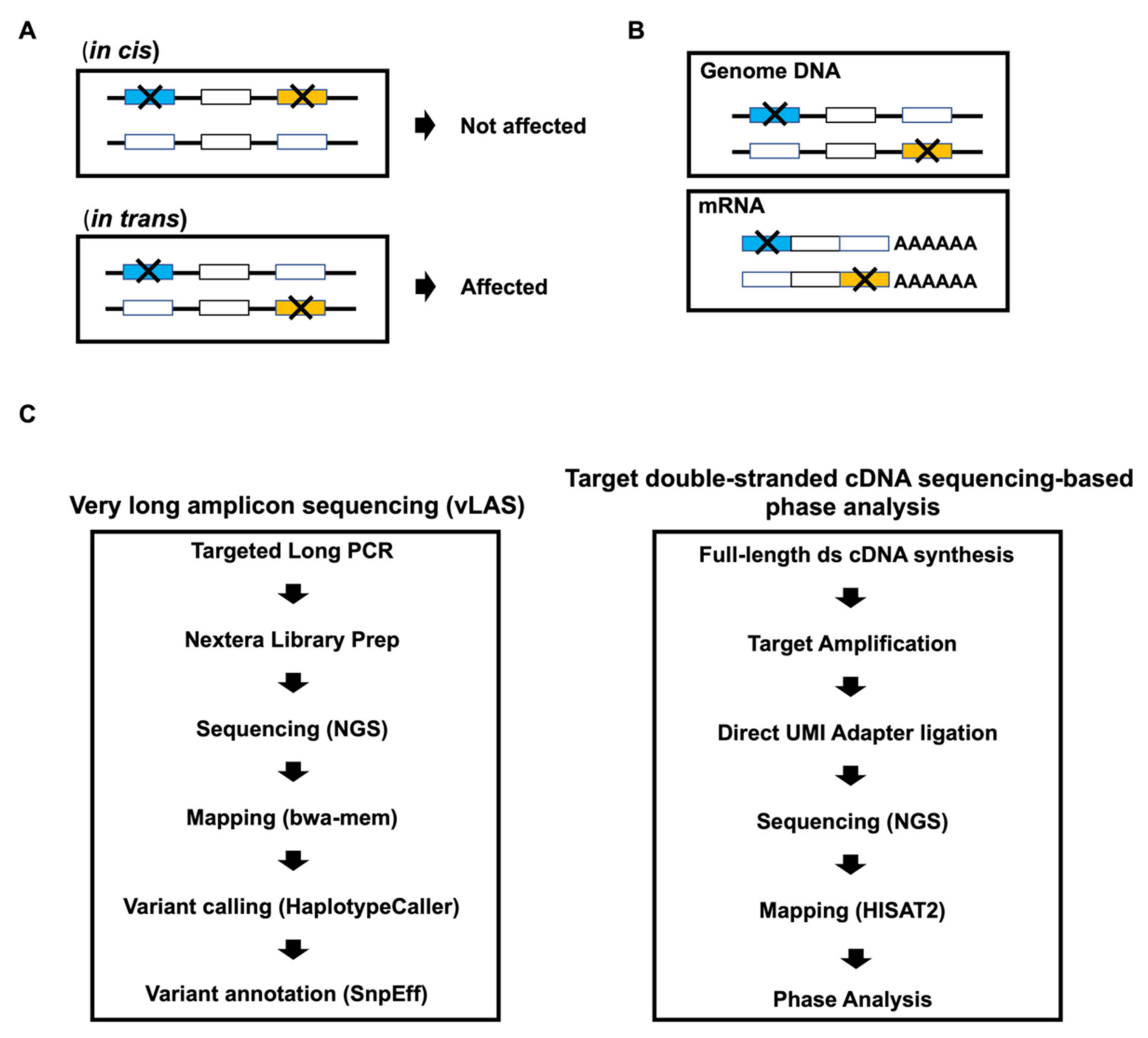
2.3. Very Long Amplicon Sequencing (vLAS)
2.4. Total RNA Extraction and Full-Length Double-Stranded cDNA Synthesis
2.5. Library Preparation for Targeted Double-Stranded cDNA Based Sequencing
2.6. Next Generation Sequencing
2.7. Data Analysis
2.8. Phase Analysis
2.9. CHIPS and Sanger Sequencing
3. Results
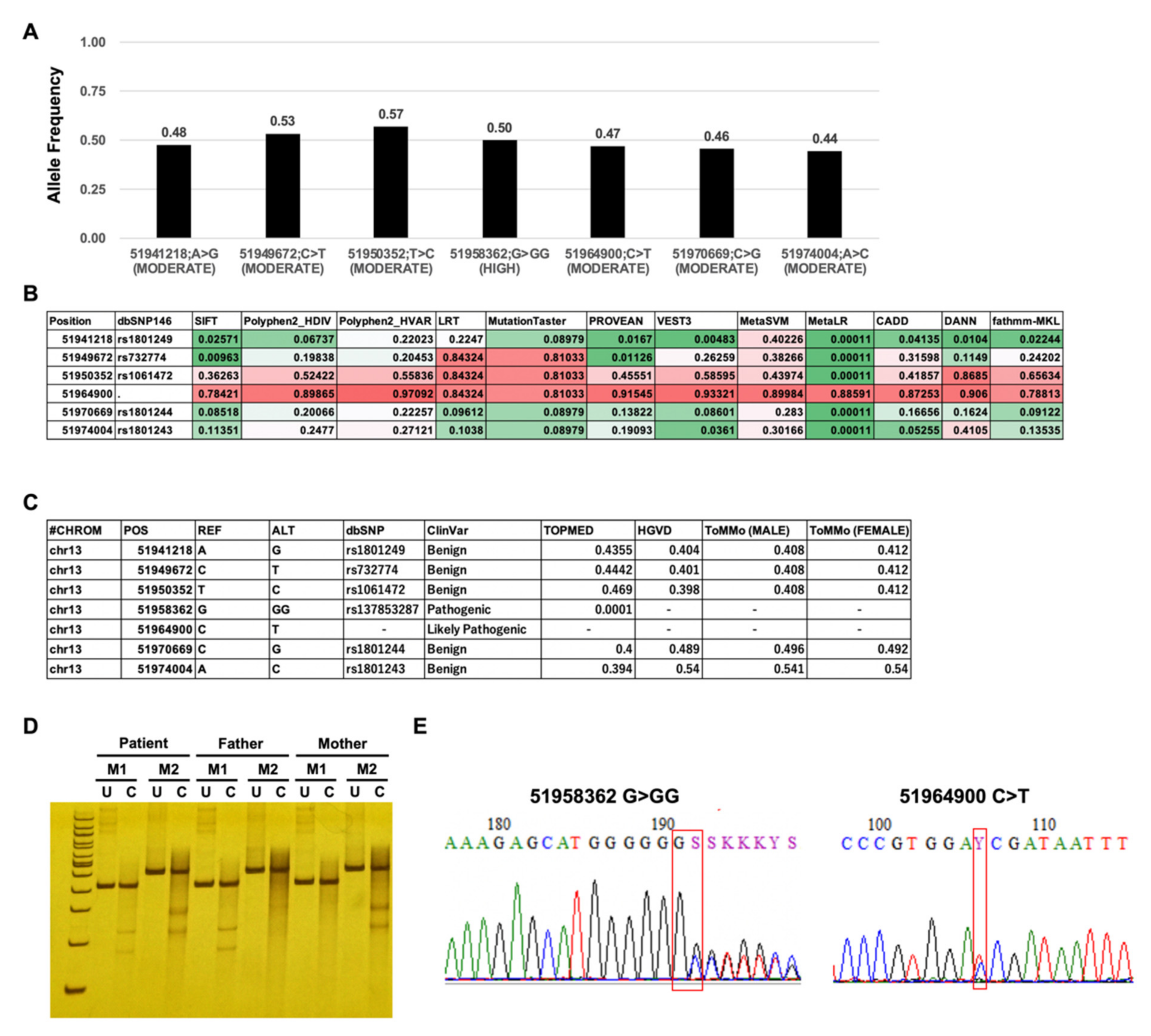
3.1. Screening for Pathogenic Mutations of ATP7B
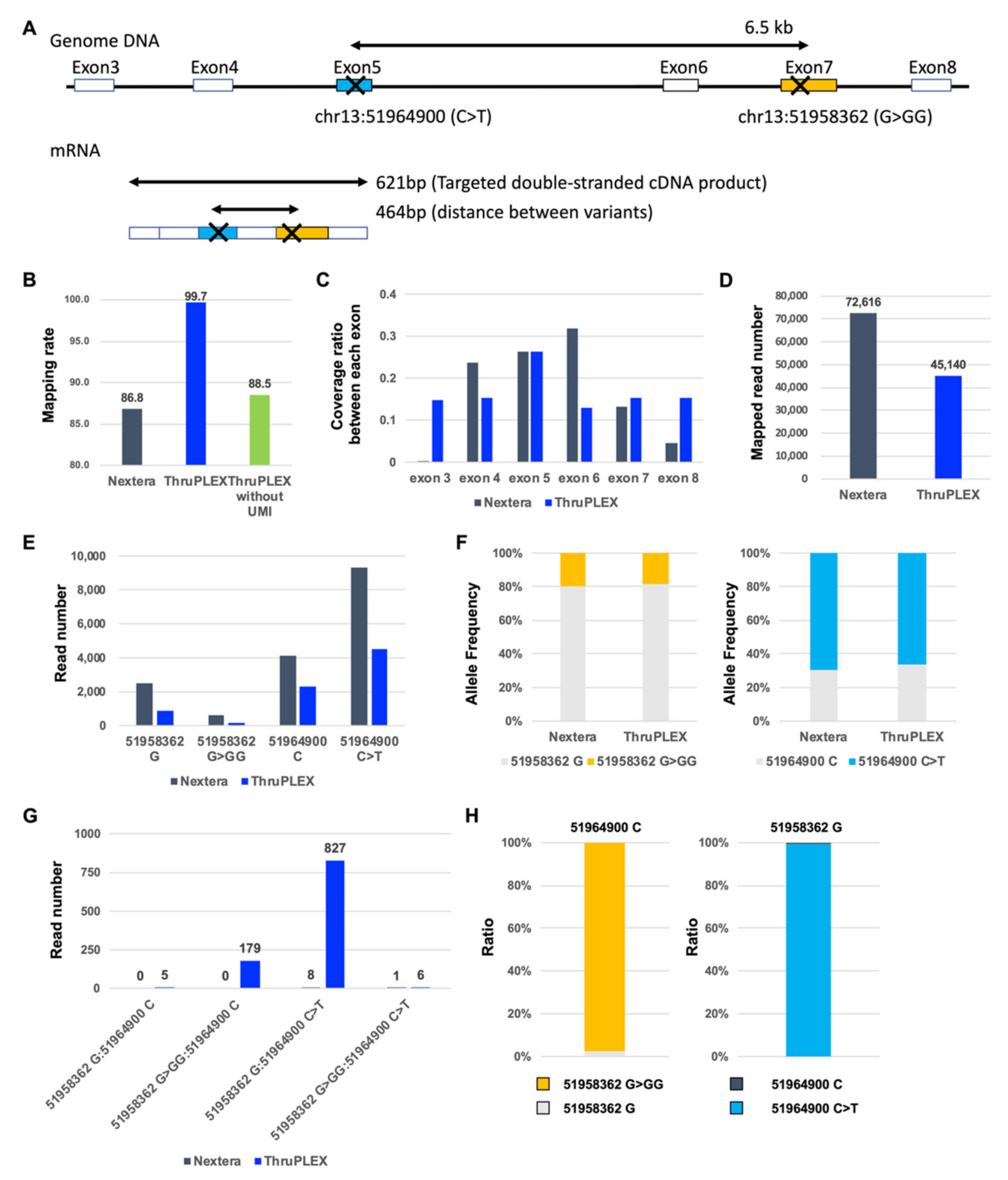
3.2. Detection of Compound Heterozygous Mutations by Targeted Double-Stranded cDNA Sequencing-Based Phase Analysis
3.3. Validation of in trans Compound Heterozygous Mutation by Trio Analysis
3.4. Frameshift Mutation Causes Differential Allelic Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartman, P.; Beckman, K.; Silverstein, K.; Yohe, S.; Schomaker, M.; Henzler, C.; Onsongo, G.; Lam, H.C.; Munro, S.; Daniel, J.; et al. Next generation sequencing for clinical diagnostics: Five year experience of an academic laboratory. Mol. Genet. Metab. Rep. 2019, 19, 100464. [Google Scholar] [CrossRef] [PubMed]
- Voelkerding, K.V.; Dames, S.; Durtschi, J.D. Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: A paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology. J. Mol. Diagn. 2010, 12, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin. Biochem. Rev. 2011, 32, 177–195. [Google Scholar] [PubMed]
- Yang, W.Y.; Hormozdiari, F.; Wang, Z.; He, D.; Pasaniuc, B.; Eskin, E. Leveraging reads that span multiple single nucleotide polymorphisms for haplotype inference from sequencing data. Bioinformatics 2013, 29, 2245–2252. [Google Scholar] [CrossRef]
- Bansal, V.; Bafna, V. HapCUT: An efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics 2008, 24, i153–i159. [Google Scholar] [CrossRef]
- Delaneau, O.; Howie, B.; Cox, A.J.; Zagury, J.F.; Marchini, J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 2013, 93, 687–696. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Fan, J.; Hu, J.; Xue, C.; Zhang, H.; Susztak, K.; Reilly, M.P.; Xiao, R.; Li, M. ASEP: Gene-based detection of allele-specific expression across individuals in a population by RNA sequencing. PLoS Genet. 2020, 16, e1008786. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Zhang, R.; Li, X.; Smith, K.S.; Knowles, D.A.; How Tan, M.; Piskol, R.; Lek, M.; Snyder, M.; Macarthur, D.G.; et al. Allelic expression of deleterious protein-coding variants across human tissues. PLoS Genet. 2014, 10, e1004304. [Google Scholar] [CrossRef]
- Miller, J.N.; Pearce, D.A. Nonsense-mediated decay in genetic disease: Friend or foe? Mutat. Res. Rev. Mutat. Res. 2014, 762, 52–64. [Google Scholar] [CrossRef]
- Frischmeyer, P.A.; Dietz, H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999, 8, 1893–1900. [Google Scholar] [CrossRef]
- Snyder, M.W.; Adey, A.; Kitzman, J.O.; Shendure, J. Haplotype-resolved genome sequencing: Experimental methods and applications. Nat. Rev. Genet. 2015, 16, 344–358. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Porubsky, D.; Garg, S.; Sanders, A.D.; Korbel, J.O.; Guryev, V.; Lansdorp, P.M.; Marschall, T. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun. 2017, 8, 1293. [Google Scholar] [CrossRef]
- Laehnemann, D.; Borkhardt, A.; McHardy, A.C. Denoising DNA deep sequencing data-high-throughput sequencing errors and their correction. Brief. Bioinf. 2016, 17, 154–179. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Patterson, M.; Marschall, T.; Pisanti, N.; van Iersel, L.; Stougie, L.; Klau, G.W.; Schönhuth, A. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J. Comput. Biol. 2015, 22, 498–509. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Schnabel, B. DNA isolation by a rapid method from human blood samples: Effects of MgCl2, EDTA, storage time, and temperature on DNA yield and quality. Biochem. Genet. 1993, 31, 321–328. [Google Scholar] [CrossRef]
- Togi, S.; Ura, H.; Niida, Y. Optimization and Validation of Multi-modular Long-range PCR-based Next-Generation Sequencing Assays for Comprehensive Detection of Mutation in Tuberous Sclerosis Complex. J. Mol. Diagn. 2021. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinf. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinf. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Niida, Y. Dual Deep Sequencing Improves the Accuracy of Low-Frequency Somatic Mutation Detection in Cancer Gene Panel Testing. Int. J. Mol. Sci. 2020, 21, 3530. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Niida, Y. Target-capture full-length double-strand cDNA sequencing for alternative splicing analysis. RNA Biol. 2021, 1–8. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Higasa, K.; Miyake, N.; Yoshimura, J.; Okamura, K.; Niihori, T.; Saitsu, H.; Doi, K.; Shimizu, M.; Nakabayashi, K.; Aoki, Y.; et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J. Hum. Genet. 2016, 61, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Tadaka, S.; Saigusa, D.; Motoike, I.N.; Inoue, J.; Aoki, Y.; Shirota, M.; Koshiba, S.; Yamamoto, M.; Kinoshita, K. jMorp: Japanese Multi Omics Reference Panel. Nucleic Acids Res. 2018, 46, D551–D557. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Niida, Y.; Ozaki, M.; Inoue, M.; Takase, E.; Kuroda, M.; Mitani, Y.; Okumura, A.; Yokoi, A.; Fujita, S.; Yamada, K. CHIPS for genetic testing to improve a regional clinical genetic service. Clin. Genet. 2015, 88, 155–160. [Google Scholar] [CrossRef]
- Woimant, F.; Poujois, A.; Bloch, A.; Jordi, T.; Laplanche, J.L.; Morel, H.; Collet, C. A novel deep intronic variant in ATP7B in five unrelated families affected by Wilson disease. Mol. Genet. Genom. Med. 2020, 8, e1428. [Google Scholar]
- Chen, Y.C.; Yu, H.; Wang, R.M.; Xie, J.J.; Ni, W.; Zhang, Y.; Dong, Y.; Wu, Z.Y. Contribution of intragenic deletions to mutation spectrum in Chinese patients with Wilson’s disease and possible mechanism underlying ATP7B gross deletions. Parkinson. Relat. Disord. 2019, 62, 128–133. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hon, T.; Mars, K.; Young, G.; Tsai, Y.C.; Karalius, J.W.; Landolin, J.M.; Maurer, N.; Kudrna, D.; Hardigan, M.A.; Steiner, C.C.; et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data 2020, 7, 399. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; Montgomery, S.B.; et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012, 335, 823–828. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ura, H.; Togi, S.; Niida, Y. Targeted Double-Stranded cDNA Sequencing-Based Phase Analysis to Identify Compound Heterozygous Mutations and Differential Allelic Expression. Biology 2021, 10, 256. https://doi.org/10.3390/biology10040256
Ura H, Togi S, Niida Y. Targeted Double-Stranded cDNA Sequencing-Based Phase Analysis to Identify Compound Heterozygous Mutations and Differential Allelic Expression. Biology. 2021; 10(4):256. https://doi.org/10.3390/biology10040256
Chicago/Turabian StyleUra, Hiroki, Sumihito Togi, and Yo Niida. 2021. "Targeted Double-Stranded cDNA Sequencing-Based Phase Analysis to Identify Compound Heterozygous Mutations and Differential Allelic Expression" Biology 10, no. 4: 256. https://doi.org/10.3390/biology10040256
APA StyleUra, H., Togi, S., & Niida, Y. (2021). Targeted Double-Stranded cDNA Sequencing-Based Phase Analysis to Identify Compound Heterozygous Mutations and Differential Allelic Expression. Biology, 10(4), 256. https://doi.org/10.3390/biology10040256