1. Introduction
The acceleration and spread of bacterial antibiotic resistance have enabled phage therapy, the use of bacteriophages (phages) as therapeutics for resistant bacterial infections, to develop and mature as a technology over the last two decades. Clinical trials are in progress in both the U.S. and Europe [
1] and procedures to access compassionate use of phage therapy are already in place in many countries [
2].
A potential hindrance to the large-scale use of phage therapy compared to traditional antibiotics is the high host specificity of phages. Traditional antibiotics can often target and treat entire classes of non-resistant bacteria, whereas phages are generally limited to a smaller subset of closely related strains [
3]. Thus, phage therapy is currently highly individualised and requires identification of the pathogenic strain and subsequent selection of a suitable phage strain. However, advancements in the field of molecular biology are enabling researchers to edit genomes more efficiently than ever. Hence, there is a potential to design and engineer recombinant phages with new phenotypic properties, such as an altered or expanded host tropism.
A variety of methodologies have been developed for genetic engineering, many of which rely on the well-established principles of homologous recombination. Possible phage genome editing approaches include CRISPR-Cas, whole-genome synthesis, and genome assembly in yeast or in cell-free TX-TL systems [
4].
The phage specificity is primarily determined by the receptor-binding proteins (RBPs) located on either tail fibres or tail spikes. These RBPs are the main focus of phage engineering, aiming to alter or expand the host tropism. A modular approach was employed by Ando et al. [
5] to reprogram wild-type
Escherichia coli (
E. coli) phage T7 to infect different bacterial species, including
Yersinia and
Klebsiella. This approach included the synthesis of the entire phage genome followed by Gibson assembly and expression in yeast cells [
5]. Demonstrating a different approach, Yosef et al. [
6] extended the host range of phage T7, intended for DNA transduction and not replication, potentially targeting bacterial species that would not normally support the propagation of phage T7. Individual recombinant phage particles were engineered by homologous recombination, incorporating a specific RBP gene into their genome. The recombinant phages showed susceptibility to multiple
Klebsiella pneumoniae and
Salmonella enterica strains [
6].
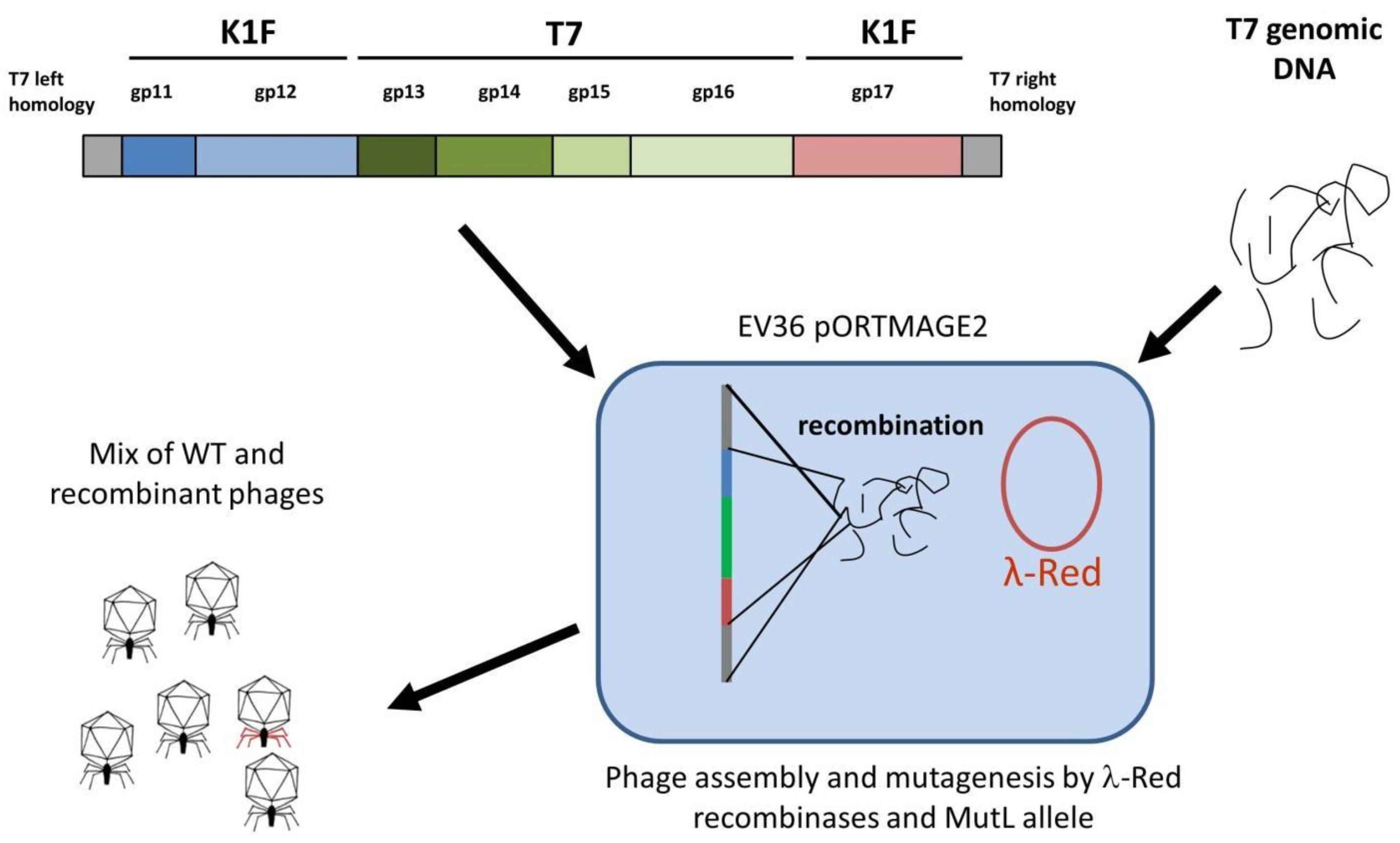
The primary goal of this work was to test multiple strategies to reprogram the host specificity of the bacteriophage T7. The ease and safety of propagating phage T7 on E. coli K12 allowed the accumulation of a vast amount of knowledge and the development of numerous techniques for this phage. This could make phage T7 an ideal candidate system to develop general phage traits (e.g., altered virulence or immunogenicity, or fusion with fluorescent labels or tags) for basic research or biotechnological purposes. Such a collection of altered T7 phages could execute specific phage-related tasks on other bacterial strains after quickly modifying host specificity. We report here the application of two different groups of strategies to reprogram the specificity of phage T7 for K1-capsule-expressing E. coli hosts. We found that if multiple phage genome segments needed to be replaced, linear-DNA mediated recombineering could yield the most promising results, even if the complete gene set required for full phage viability was unknown.
3. Results
In this project, we tested multiple strategies to alter the host specificity of phage T7, to provide the capability of infecting K1-capsule-expressing
E. coli cells. As a starting point, we analysed the growth and plaque-forming abilities of phage T7 and K1F on
E. coli MG1655 and
E. coli EV36 strains displaying the K12 and K1 capsules, respectively. The results, detailed in
Table 1, indicated that the liquid-culture-mediated growth of T7 and K1F is only possible on
E. coli MG1655 and EV36 strains, respectively. However, plaque formation in similar numbers was observed when switching the bacterial hosts (i.e., T7 on EV36 and K1F on MG1655), albeit yielding smaller plaque diameters. This indicated that the host specificities of the phages were not completely exclusive at the starting point of our experiments.
In the case of phage T7, binding to the cell surface is ensured by the six tail fibres. Each tail fibre consists of three Gp17 proteins, all attached to the phage tail with their N-termini. The phage tail is made up of a dodecamer of the Gp11 and a hexamer of the Gp12 proteins. Gp7.3, an essential protein that is injected into the target cell, is also present in the tail in about 30 copies at an unknown location [
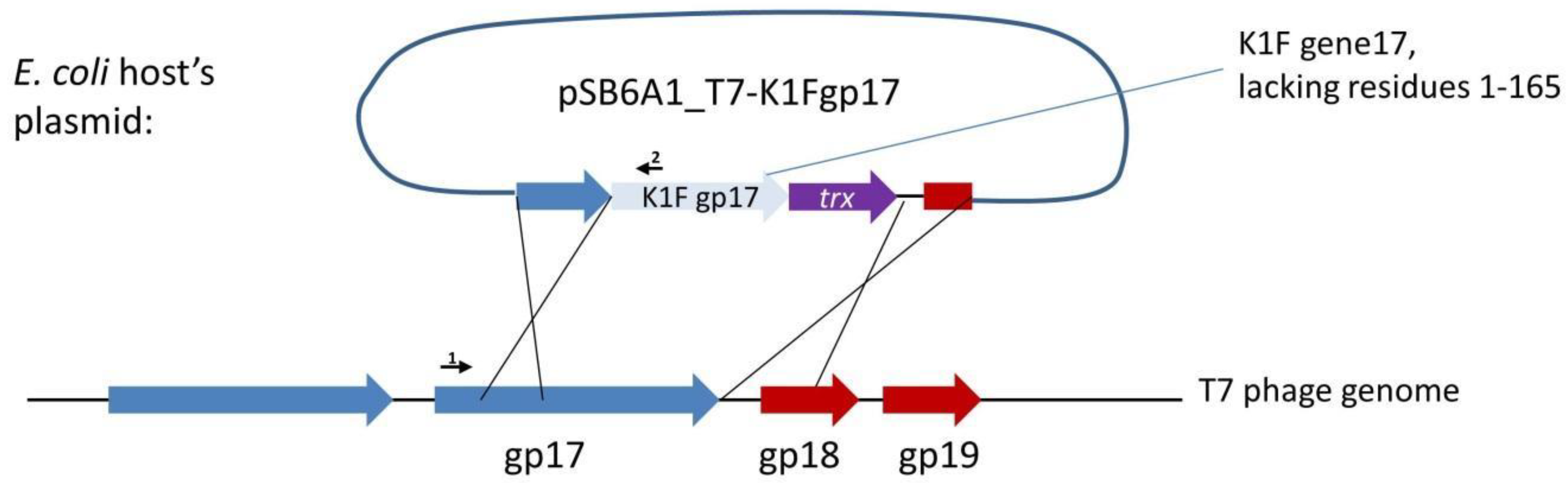
18]. Our first strategy to completely switch the specificity of phage T7 to K1 hosts targeted the tail fibre using plasmid-mediated phage engineering, i.e., the growth of T7 on
E. coli K12 cells harbouring a donor plasmid (pSB6A1_T7-K1Fgp17) that carries a modified
gp17 gene (Strategy I). This plasmid was designed to include the 860 C-terminal residues of the K1F
gp17 (possessing endosialidase activity), thioredoxin (
trxA) for positive selection, and two flanking phage T7 homology regions (
Figure 2). Sequence similarities between phage K1F and phage K1-5, another K1 capsule-targeting phage, showed that the 204 amino acids (AAs) of the N-terminal end of K1F Gp17 were not necessary for the correct folding of the enzymatic protein. We, therefore, designed homologies to obtain a fusion protein where the 165 AA N-terminal sequence of K1F
gp17 is replaced with the 162 AA N-terminal end of T7
gp17.
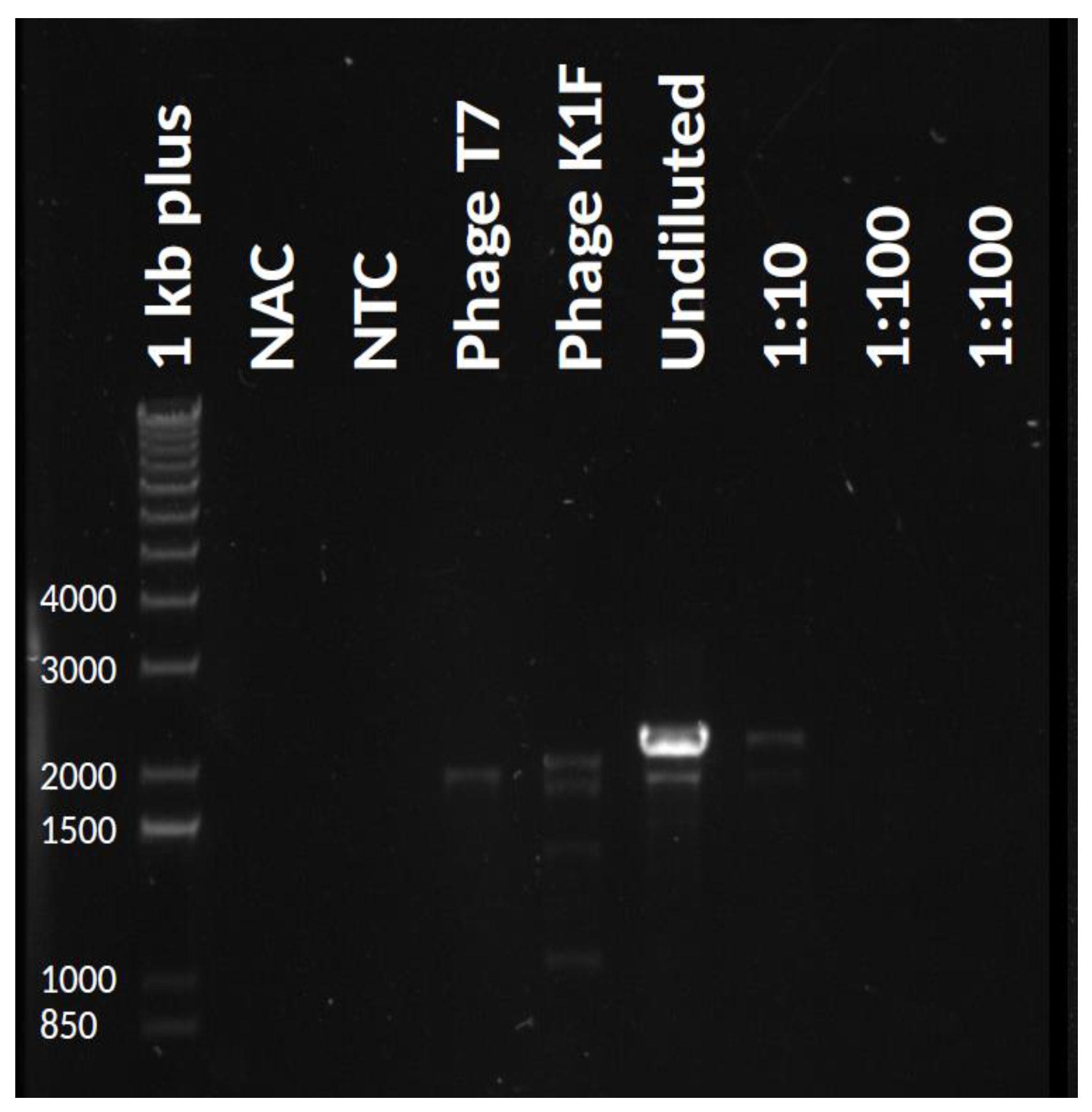
Propagation of phage T7 on
E. coli K-12 (MG1655)/pSB6A1_T7-K1Fgp17 yielded a phage lysate that contained detectable amounts of the fusion
gp17 gene, as verified by PCR using primers AS072 and AS081 (
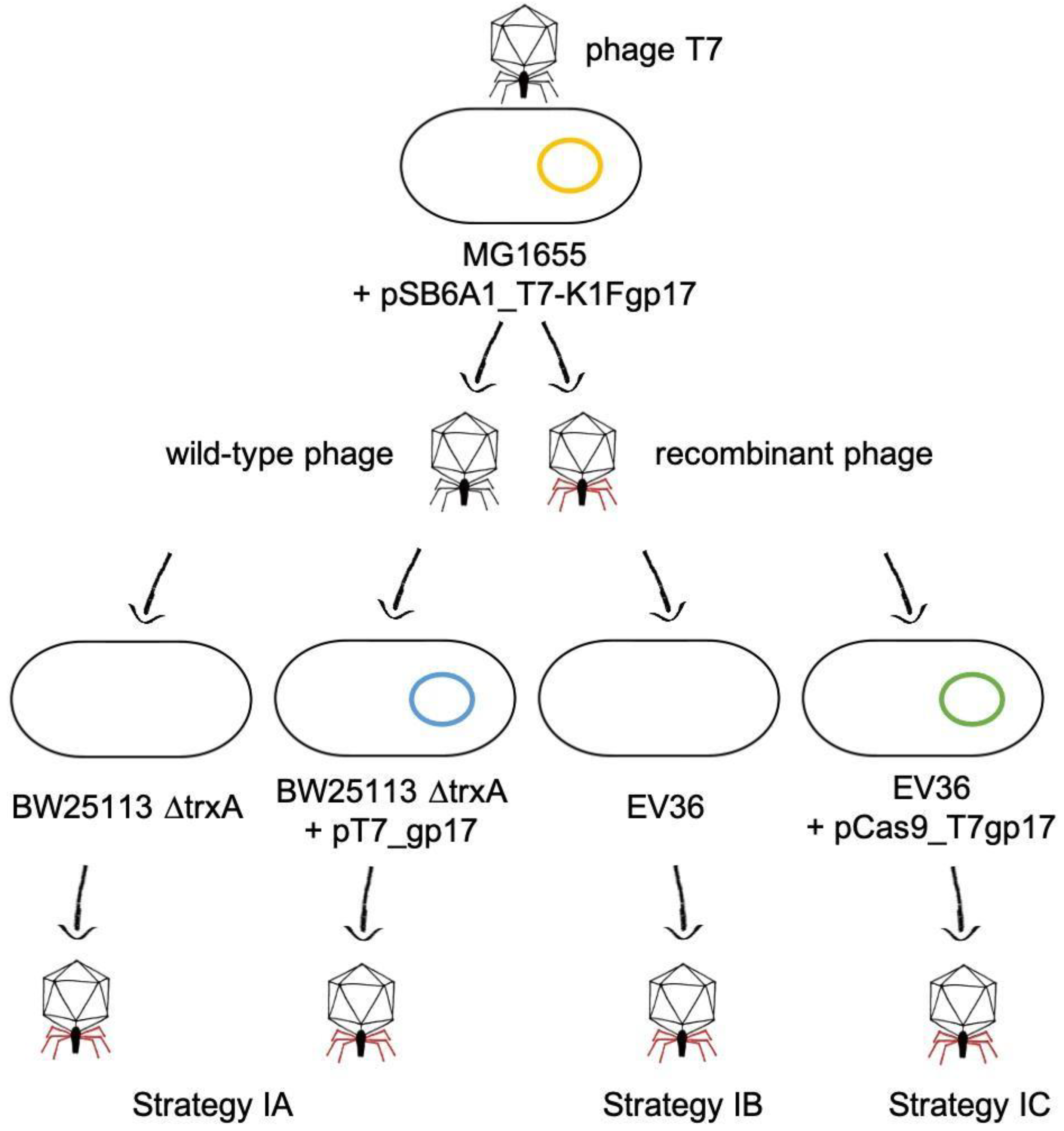
Figure 3). We applied three different strategies (IA-C) to select for or enrich the recombinants within the phage mix (
Figure 4). Strategy IA consisted of phage growth on
E. coli BW25113 ∆
trxA, which lacks the thioredoxin gene. Since this gene is essential for phage replication, this host only allows the replication of phage genomes that have acquired a copy of the
trxA gene. Unfortunately, PCR-detectability of the chimeric phage was lost over the first few rounds of phage propagation. The failure of this method can be, in part, attributed to the fact that our ∆
trxA strain did not carry a K1 capsule and was, therefore, a suboptimal host for the propagation of the recombinant phage. Attempting to boost phage propagation by providing the Gp17 protein of T7 in trans did not solve this issue: the PCR signal also disappeared when growing the phages on
E. coli BW25113 ∆
trxA + pT7_gp17.
Strategy IB was more straightforward; it relied on the potentially improved ability of the recombinant phage to lyse liquid cultures of
E. coli EV36, a K1 capsule-expressing host. We propagated the phage mix on liquid cultures of
E. coli EV36, measuring phage titers after every round of phage growth (even if no visible lysis occurred) using plated EV36. Overall, the phages displayed a weak and poorly reproducible ability to grow in liquid EV36 cultures. As apparent from
Table 2, the titers measured in the course of three passages strongly fluctuated; furthermore, during the first and third steps, we only recovered the phages that we had mixed with the cells, indicating negligible phage propagation. A very similar titer pattern was observed when starting not with the complete phage lysate, but with a plaque obtained by plating the lysate on
E. coli EV36. Importantly, in both series of propagations, the phage mix lost the PCR-positivity by the third passage. Overall, we concluded that propagating the phage mix on a K1-expressing host did not select for the recombinant phages and could not even maintain the fusion
gp17 genotype within the mix.
In Strategy IC, we intended to further increase the pressure enriching recombinant phages by applying CRISPR-Cas selection. We grew the phage mix on K1 capsule-expressing hosts that carried the S. pneumoniae CRISPR-Cas machinery targeted against either of two loci in the T7 gp17 gene, not present in the fusion gp17. Growth of the phage mix on E. coli EV36/pCas9_T7gp17 or EV36/pCas9_T7gp17-2 was carried out both in liquid culture and on plates, as described in the Methods. In both cases, the PCR-positivity of the obtained phages was lost already after the first round of phage growth, irrespective of the guide RNA used.
At this point, it became evident that the recombinant phage was not viable, or at least its selective disadvantage compared to T7 was greater than the selective pressure exerted by any of the three systems described above. A potential explanation and solution were provided by the work of Ando et al. [
5], who made similar observations when replacing the
gp17 gene of T7 with that of
Klebsiella phage K11. In their work, the recombinant phages were not viable unless
gp11 and
gp12 (the genes encoding the adaptor and the nozzle proteins of the phage tail, respectively) were also replaced by their K11 counterparts. The authors explained this requirement by the fact that the
gp17-encoded tail fibres attach to the interface between the Gp11 and Gp12 proteins. In light of this information, we redesigned the donor plasmid to comprise the K1F derived
gp11,
gp12, and
gp17 genes, as well as the T7-derived
gp13, gp14, gp15, and
gp16 genes, as shown in
Figure 1. The gene set was to be flanked by two homologous ends allowing its entry into the T7 genome to replace the native
gp11-
gp17 segment.
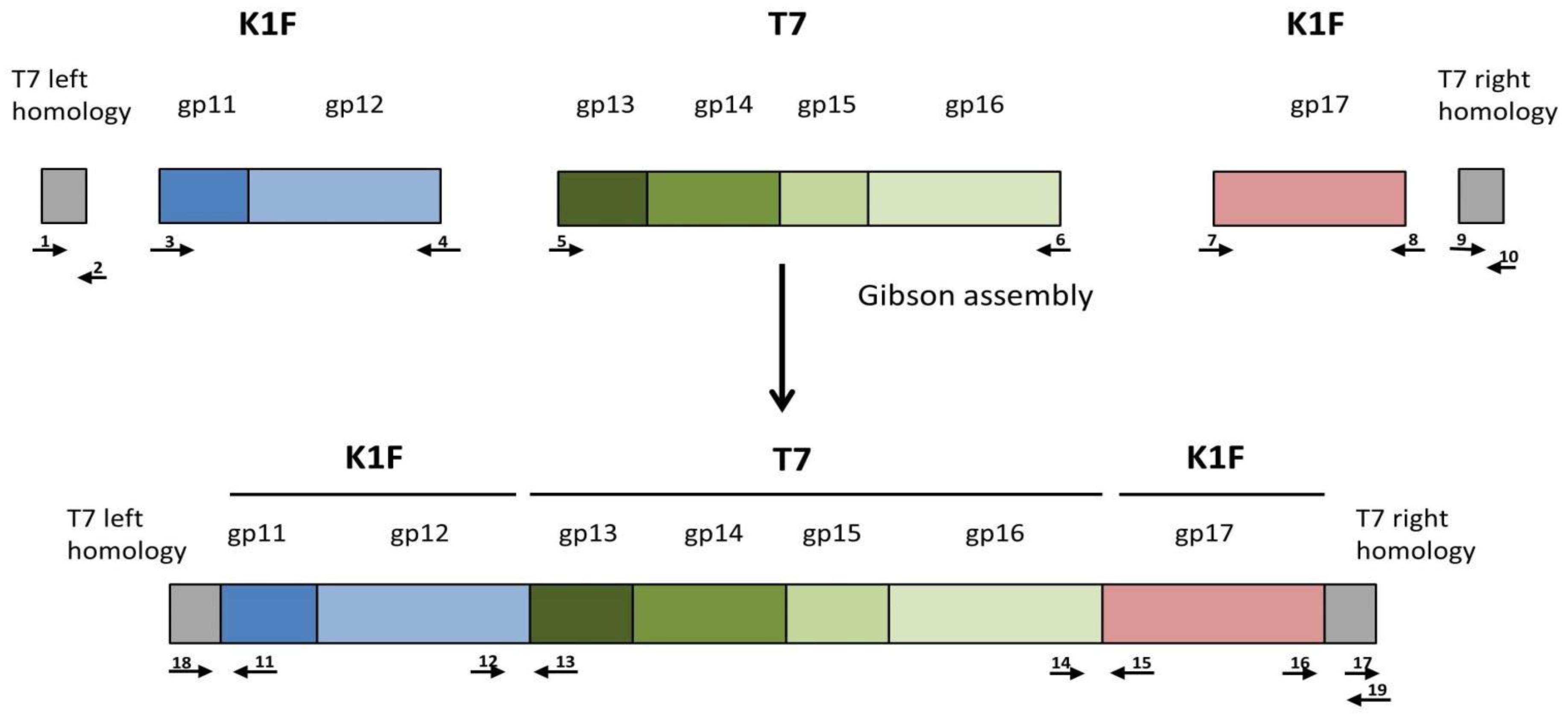
The donor plasmid was to be constructed as a four-way assembly (T7 left homology +
gp11-
12;
gp13-16;
gp17 + T7 right homology; bacterial artificial chromosome segment) (
Figure S1) using the method of Gibson et al. [
19]. Gel electrophoresis of the reaction product and PCR of the assembly joints indicated that the 14.2 kbp-long linear cassette was most likely assembled (
Figure S2). However, cloning it into pBeloBAC11 was not successful, irrespective of whether
E. coli MDS42, MG1655, or EV36 was used as a host, possibly due to the toxicity of the construct. We, therefore, abandoned plasmid-mediated phage editing and switched to Strategy II (
Figure 5), which relies on bacteriophage recombineering using electroporated DNA (BRED) [
20]. This strategy builds on the fact that if purified phage DNA is electroporated into an
E. coli cell expressing the λ-Red recombinases, complete phage genomes are assembled, yielding phage plaques within the bacterial lawn. If a linear DNA fragment with appropriate homology arms is included in the electroporation mix, a certain fraction of the obtained plaques will contain recombinant phages, detectable by plaque-PCR.
For strategy IIA,
E. coli EV36/pORTMAGE2 cells were electroporated with a mix of T7 genomic DNA and the linear DNA fragment, as shown in
Figure 5. After plating and overnight growth, a total of 14 large (>10 mm) clear plaques were observable. We analysed all plaques by four distinct PCR reactions (listed in Materials and Methods), screening for the presence of the four T7/K1F joints. We found that all of the plaques contained at least one of the two transgenic segments (i.e.,
gp11-12 or
gp17) and four of them contained both integrated into the T7 genome at the correct locus (
Figure S3). Interestingly, small plaques formed by wild-type T7 did not emerge (otherwise seen when the phage genome is transformed without linear DNA). A double-positive plaque was picked, and the phages were recovered and further propagated on liquid
E. coli EV36 culture to verify its growth ability and to enrich for virions containing the altered tail fibre. No lysis was observable in the liquid medium, and the obtained plaques displayed the absence of
gp17 and
gp11-12 genes, as analysed by PCR. As an alternative (Strategy IIB), the phages recovered from the plaque were grown on
E. coli EV36/pORTMAGE2 to provide further possibilities of recombination (by the λ-Red recombinases) and an increased rate of point mutations (by the
mutL allele). Growth on liquid
E. coli EV36/pORTMAGE2 culture was possible, as the cells were consistently lysed. However, the transgenic segments of
gp17 and
gp11-12 were lost in the first and second round of phage growth, respectively. Genes of the T7 phage were readily detectable by PCR in all rounds of phage growth (
Figure S4). The continued lysis of the non-corresponding host by phage T7 is explained by the presence of λ-Red recombinases. The expression of λ-Red in bacterial cells has recently been shown to provide sufficient genetic reorganisation to lyse non-canonical hosts [
21] (in a control experiment, we found that serial propagation of T7 on
E. coli EV36/pORTMAGE2 liquid cultures led to full lysis starting from the second round, without any linear DNA fragment added; data not shown). The obtained phages nevertheless retained their ability to lyse
E. coli MG1655, as well. The activity of the K1F
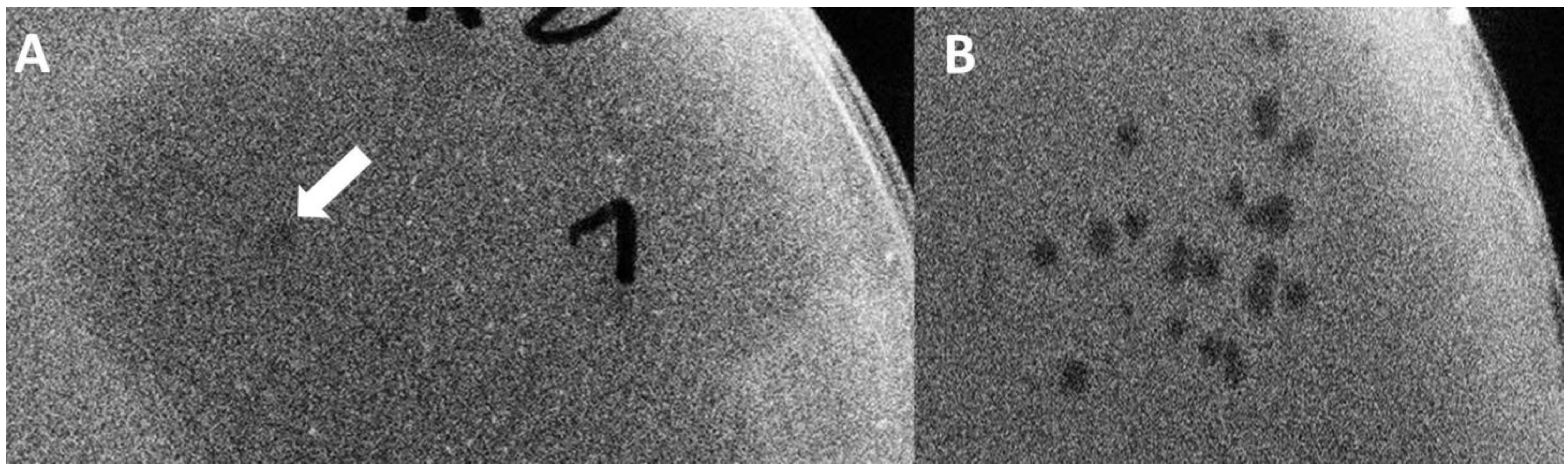
gp17 was still present after the end of the serial phage growth experiment in the form of two-stage plaques (
Table 3,
Figure 6), assuming some lasting genetic change did occur. The same could not be seen when wild-type T7 was assembled without K1F gene fragments.
Finally, in Strategy IIC, we repeated the BRED, but instead of plating the transformed bacterial mix and picking a plaque, we grew the bacteria (and the emerging phages) directly in a liquid culture. This avoids the bottleneck introduced by randomly picking a plaque and allows the competition of all putative recombinant and wild-type T7 phages within a single mix. Growth took place without antibiotic selection to promote the quick loss of the pORTMAGE2 plasmid. In this case, although bacterial lysis was not always visible in the successive rounds of growth, phages were always detectable, despite the lack of pORTMAGE2. However, transgenic fragments were quickly lost from the phage genome. In all versions of Strategy II, the obtained plaques displayed a two-stage phenotype, usually starting from the second or third round of growth. This meant a small, clear centre surrounded by a turbid halo assuming the endosialidase activity of the K1F Gp17 protein [
22] and the area of both kept increasing day-by-day (
Figure 6). The observations made with BRED experiments are summarised in
Table 3.
4. Discussion
Ever since the discovery of bacteriophages, knowledge concerning their host range has been vital for biomedical applications for at least two reasons. On the one hand, phage typing has long been used in microbial diagnostics to identify and classify the pathogens isolated from bacterial infections [
23]. On the other hand, phage therapy requires the administration of phages capable of lysing the targeted pathogen. These needs explain the increased attention paid towards phages that display a broad host range [
24] or a host range dependent on environmental factors, like temperature [
25]. Later, spontaneously arising host range mutants were often described [
26,
27], and with the advent of DNA sequencing, the mutations responsible for the changes were also successfully identified [
28,
29]. With the accumulation of sequencing data, comparative genomic studies of phage sequences became possible. These revealed that in certain cases, exchanges of large genomic segments are responsible for host switching [
30]. Such information, along with the development of phage engineering techniques, allowed the reprogramming of the host range of certain phages by replacing host-range-determinant DNA segments [
5,
31]. Recently, the structure-guided design of receptor-binding proteins has proven to offer an even more advanced method of redirecting phages towards new hosts [
32].
Our work fits into this series by providing an example of transferring one or more large genomic segments between phages to alter the host tropism of the recipient phage. In the course of this project, we tested two groups of strategies and two different genetic constructs. Strategy I applied plasmid-based phage editing using either trxA (IA), host-specificity (IB), or CRISPR-Cas-based selection (IC). Strategy II relied on BRED, using a host-specificity-based selection of the recombinant phage. For Strategies IIA and IIB, a single positive plaque was picked for the outgrowth of the putative recombinants on cells either lacking or containing the λ-Red recombinase enzymes, respectively. Alternatively, outgrowth of the entire phage lysate was attempted in the absence of λ-Red recombinase enzymes for Strategy IIC.
To the best of our knowledge, this is the first published work aiming to tune phage T7 tropism towards a K1-capsule-expressing host. This type of specificity-change could bear clinical relevance in the future, when the rapid redirection of certain well-established phages towards novel bacterial pathogens is required. From the molecular genetic point of view, all six strategies accomplished the goal of producing detectable recombinant constructs. Since well-established techniques were used for their selection, we attribute the differences seen in the sustainability of the constructs not to differences of the selection methods themselves but instead to the differences in the genetic design. Strategy I only altered
gp17 and left the 5′ end of the gene unchanged, while Strategy II applied the complete exchange of the
gp17 gene, along with that of
gp11 and
gp12. Since the latter strategy yielded plaques that stood closest to the phenotype of completely lysing the new host, we conclude that modification of either
gp11 or
gp12, or both, are required, in addition to that of
gp17 for the reprogramming of T7 to lyse K1-type hosts. The temporary nature of such lysis, however, indicates that these genetic changes were still not sufficient to develop a stable viable phage. Although this may be a shortcoming in some instances, it is satisfactory for numerous experimental setups. The most prominent example is the engineering of transducing phages or phage libraries, where efficient phage binding, DNA injection, and the overcoming of defence mechanisms without autonomous phage replication are enough to meet the requirements of practical applications [
6].
Besides yielding unstable reprogrammed phages, our work also provides findings that may help design future host switching experiments. First, we observed that the presence of pORTMAGE is capable of allowing the growth of T7 on
E. coli EV36 in liquid, irrespective of donor DNA. This is in line with recent observations reporting that λ-Red recombinase expression enables T7-like bacteriophages that do not normally propagate in
E. coli to be recovered following genome transfection [
21]. Including donor DNA in the transformation mix nevertheless exerted the temporary effect of yielding large clear plaques, which was eventually lost. This means that (a) the genes responsible for efficient lysis of
E. coli EV36 are present; and (b) no pure, viable recombinant phage, which could pass on its genetic composition to its offspring, was made. A mixed infection (T7 + recombinant phages) or the integration of donor DNA into the bacterial host genome probably provided a genetic background that resulted in the formation of the large clear plaques seen in the first plating of Strategy IIA. Second, the observed two-stage plaques indicated the expression of the K1F
gp17 gene [
22]. According to our hypothesis,
gp17 was transiently expressed, and Gp17 enzymatically broke down the capsule, forming the turbid halo. This allowed the access of T7 to the cell surface, which formed the clear centre of the two-stage plaques. However, the gene construct is unstable; the proteins encoded by the construct cannot assemble to yield viable phage particles alone. If a helper T7 is present, it can package the construct and transfer, but again, it will not form viable particles alone. Therefore, recombinant constructs are quickly lost from the phage mix. If the infected strain harbours λ-Red recombinases, it can protect the transformed
gp17 gene cassette and potentially integrate
gp17 transferred by the helper T7 phage into the bacterial genome. This allows transient production of endosialidase, which, upon cell lysis, will break down the capsule of neighbouring cells. The case of a mixed infection (T7 + recombinant phages) is supported by the fact that even when propagating phages derived from isolated plaques declared recombinant by PCR, wt T7 phages eventually become dominant and even exclusive after a few cycles of growth on EV36. BRED is known to produce mixed plaques [
20], since both WT and recombinant phage genomes can be assembled in the same transformed host, which warrants that wt T7 is available as a helper phage during the initial growth of the recombinants. Transferring to liquid phase, however, lowers the chances of co-infection due to the effect of dilution, leading to the eventual cessation of the packaging of the recombinant phage genome into T7-virions.
Finally, although a stable phage displaying host switching was not assembled in this work, the large clear plaques are a strong indication that the necessary genes required for serial lysis of a non-canonical host were all present. The T7 engineered phage, even though it was not stable for the long term, still presented broader tropism and could efficiently target EV36 presenting the K1 capsule, which is normally a host for the K1F phage. The use of BRED for this purpose could be a quick way to test the potential use of a new tail fibre without the need to engineer a stable phage.
Concerning the future, the information collected to this point could provide a basis for the further enhancement of the hybrid construct, ultimately leading to a stable T7-K1F chimeric phage. The two most straightforward explanations of the instability of our chimera could be either the inadequate expression of genes present on the construct or the inappropriate assembly of the encoded proteins due to their potentially incompatible adjacent surfaces. Although we cannot completely exclude the former, earlier reports on reorganizing genes within a phage genome indicated a relatively high level of robustness [
33,
34]. Perhaps more likely is the incompatibility of neighbouring protein surfaces. This would suggest that modular phage reorganization, which works quite effectively in certain cases [
5], is insufficient in others and demands sub-modular tuning for successful host-switching. The accumulation of protein structural data and the development of novel combinatorial genome editing techniques will certainly aid both the design-based and the selection-based strategies aiming to solve such issues in the future.



 and
and 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}