
Identifying Hidden Viable Bacterial Taxa in Tropical Forest Soils Using Amplicon Sequencing of Enrichment Cultures


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
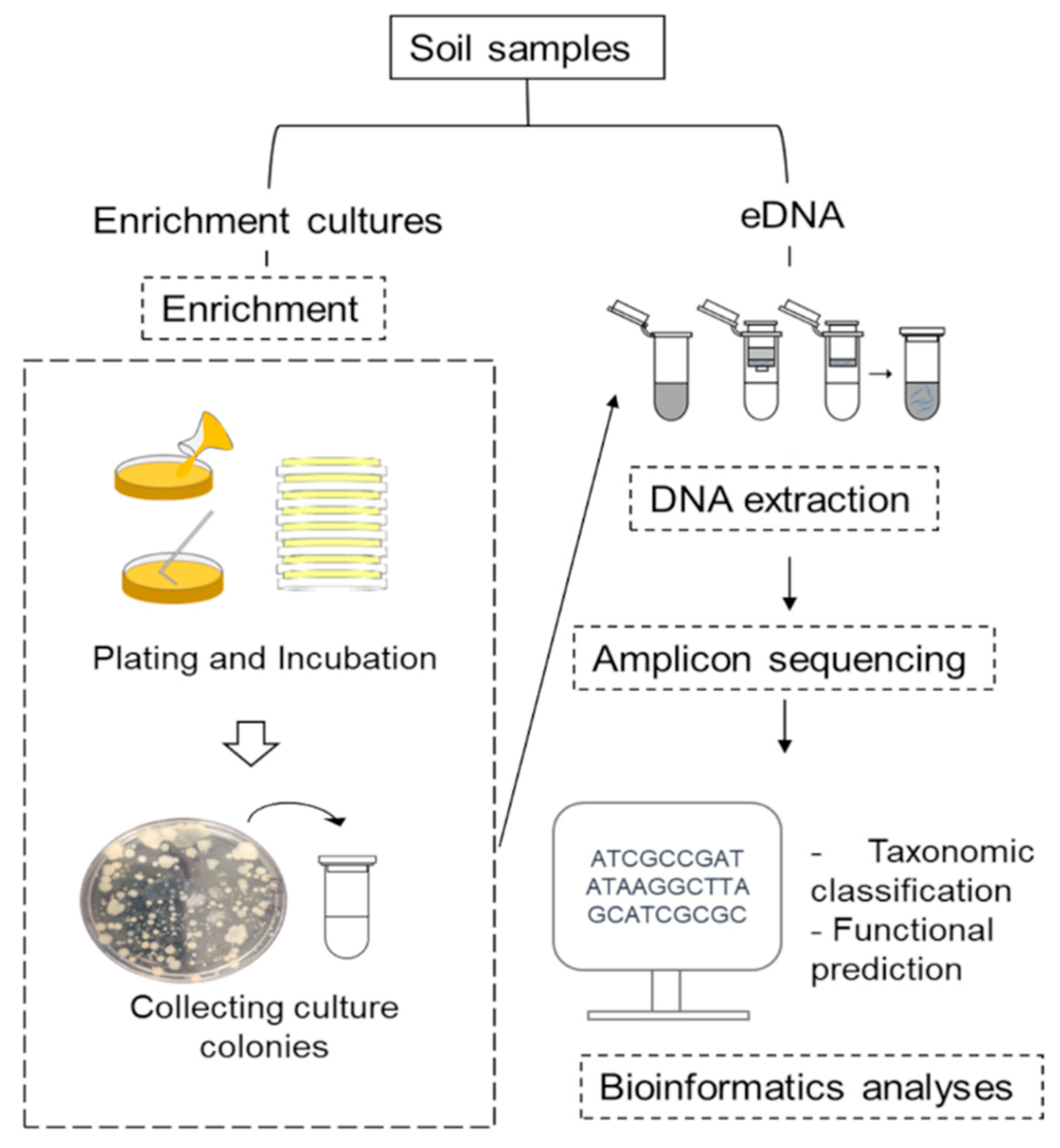
2. Materials and Methods
2.1. Soil Collection
2.2. Bacterial Enrichment
2.3. DNA Extraction, Amplicon Sequencing and Sequence Processing
2.4. Statistical Analyses
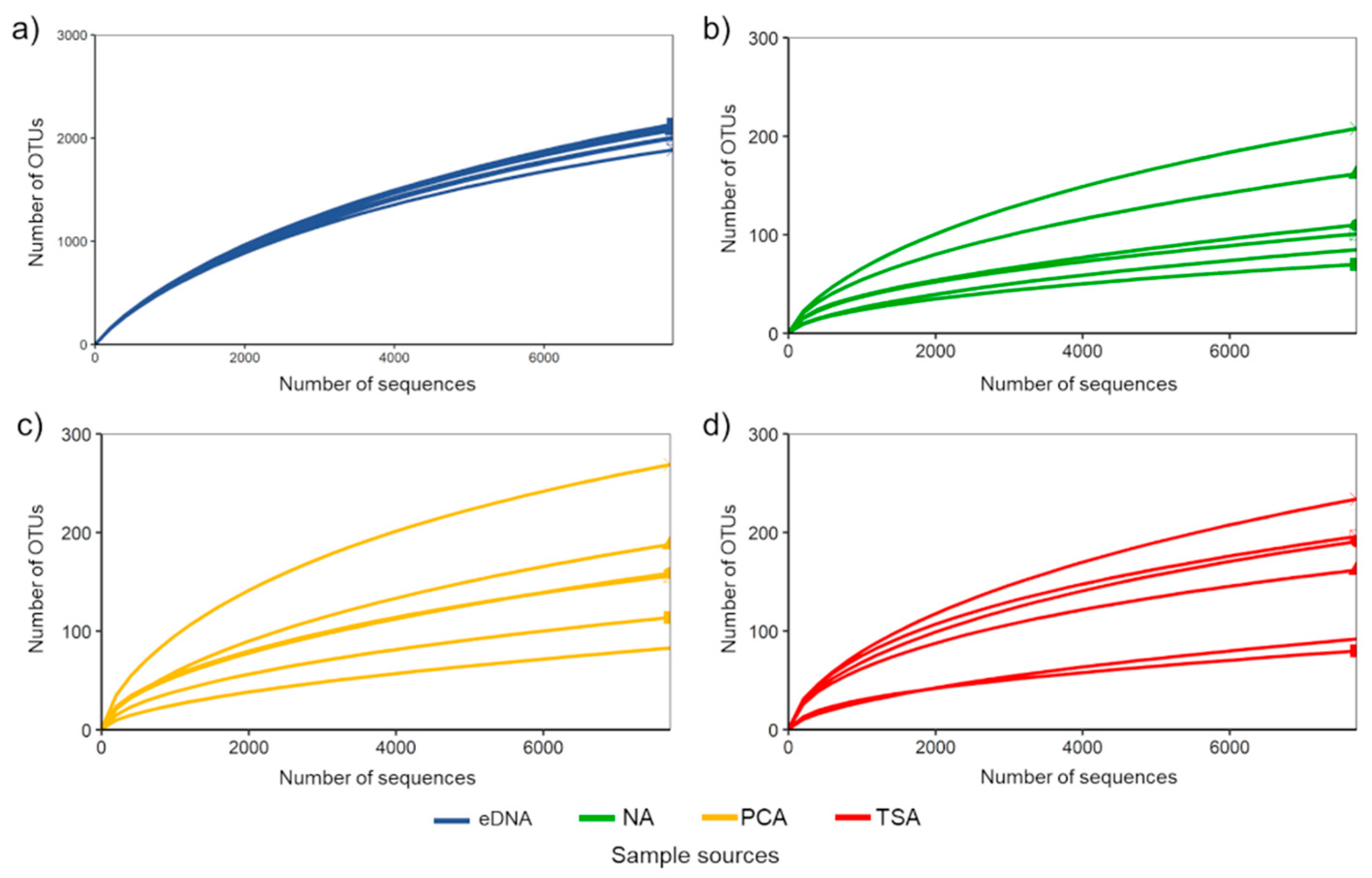
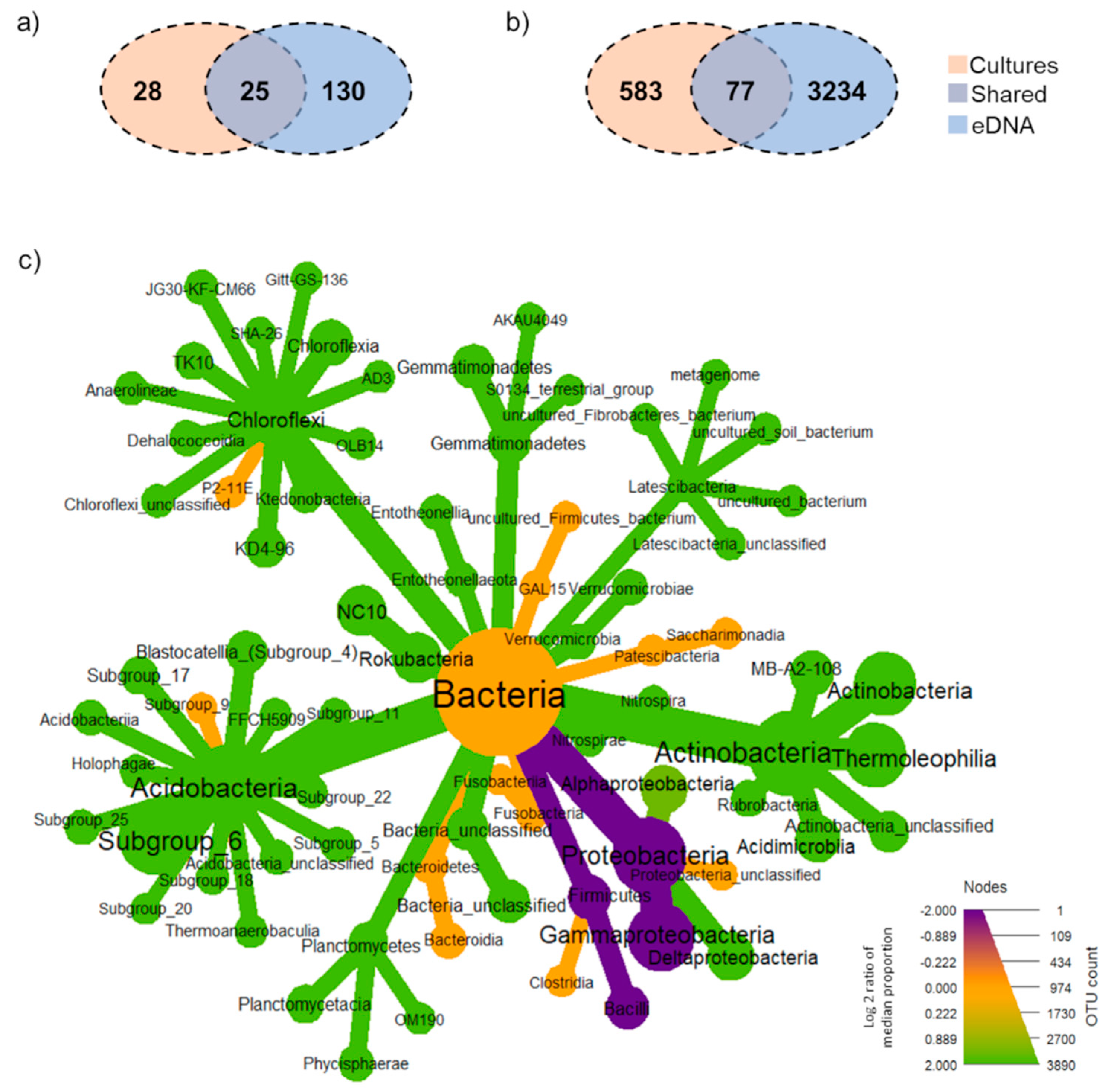
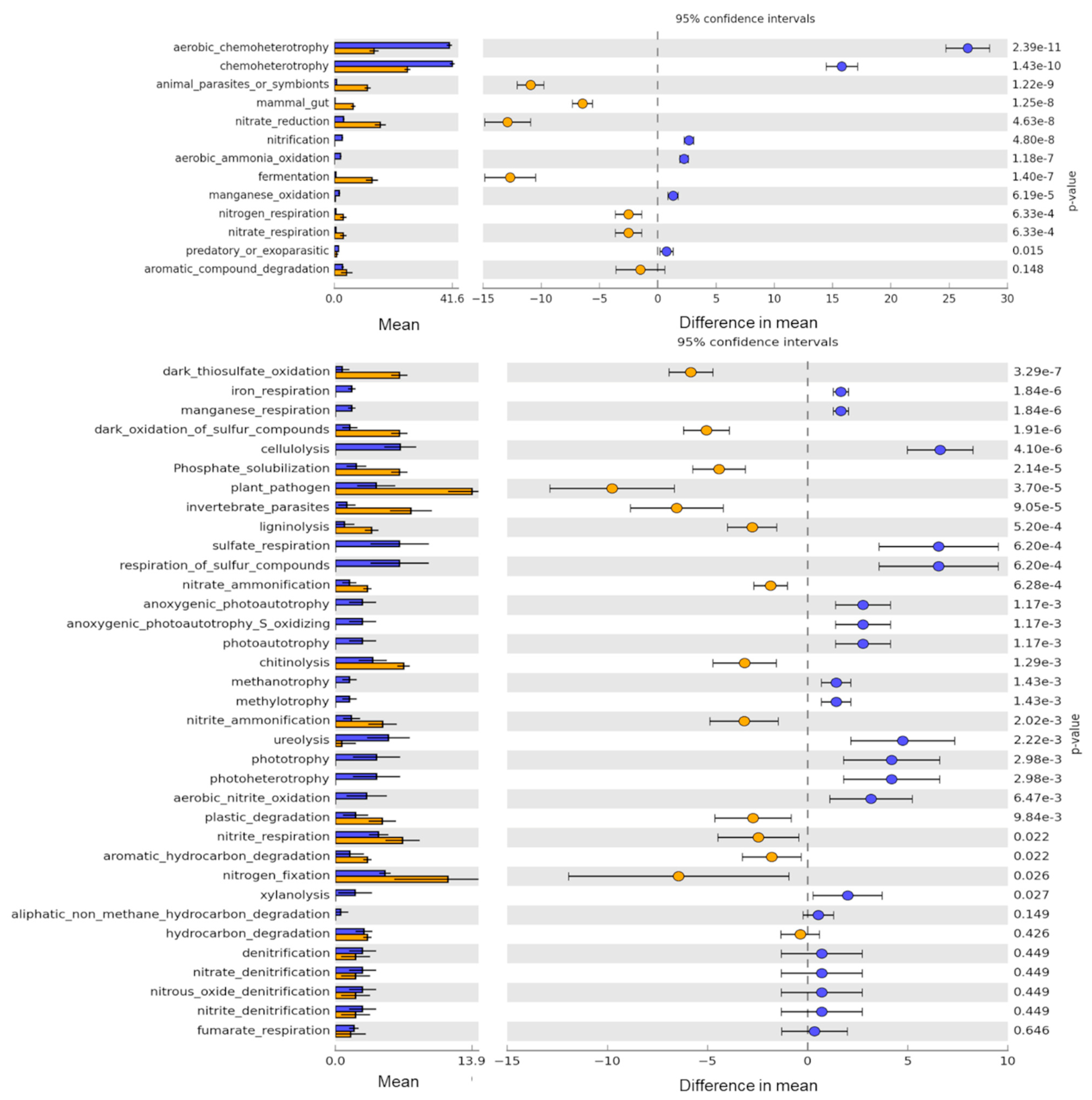
3. Results
3.1. General Overview of Bioinformatics and Taxonomic Information of Becteria Detected by Culture-Based and eDNA-Based Methods
3.2. Total Soil Bacterial Community: Unique, Overlap and Prevalent Taxa Derived from Each Method
3.3. Bacterial Taxa Derived from Different Non-Selective Media
3.4. Potential Bacterial Functions Captured by Culture-Based Method as Compared to eDNA
4. Discussion
4.1. Revealed Hidden Viable Bacterial Taxa in Tropical Forest Soils Using Amplicon Sequencing of Enrichment Cultures
4.2. Limited Number of Viable Bacteria Detected by Amplicon Sequencing of Enrichment Cultures and Possible Way to Improve the Method
4.3. Detection of Diverse Bacterial Functional Groups in Soils by Amplicon Sequencing of Enrichment Cultures
4.4. Potential Applications of Enrichment Culture-Based Amplicon Sequencing
4.5. Future Perspective on the Study of Viable Soil Microorganism and Outlook
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bodelier, P.L.E. Toward understanding, managing, and protecting microbial ecosystems. Front. Microbiol. 2011, 2, 80. [Google Scholar] [CrossRef] [Green Version]
- Graham, E.B.; Knelman, J.E.; Schindlbacher, A.; Siciliano, S.; Breulmann, M.; Yannarell, A.; Beman, J.M.; Abell, G.; Philippot, L.; Prosser, J.; et al. Microbes as engines of ecosystem function: When does community structure enhance predictions of ecosystem processes? Front. Microbiol. 2016, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alori, E.T.; Babalola, O.O. Microbial inoculants for improving crop quality and human health in Africa. Front. Microbiol. 2018, 9, 2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngalimat, M.S.; Mohd Hata, E.; Zulperi, D.; Ismail, S.I.; Ismail, M.R.; Mohd Zainudin, N.A.I.; Saidi, N.B.; Yusof, M.T. Plant growth-promoting bacteria as an emerging tool to manage bacterial rice pathogens. Microorganisms 2021, 9, 682. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; McAuliffe, O.; Ross, R.P.; Hill, C.; O’Mahony, J.; Coffey, A. Bacteriophages and bacterial plant diseases. Front. Microbiol. 2017, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Sarsaiya, S.; Wu, Q.; Lu, Y.; Shi, J. A review of plant leaf fungal diseases and its environment speciation. Bioengineered 2019, 10, 409–424. [Google Scholar] [CrossRef] [Green Version]
- Al-Awadhi, H.; Dashti, N.; Khanafer, M.; Al-Mailem, D.; Ali, N.; Radwan, S. Bias problems in culture-independent analysis of environmental bacterial communities: A representative study on Hydrocarbonoclastic bacteria. SpringerPlus 2013, 2, 369. [Google Scholar] [CrossRef] [Green Version]
- Blagodatskaya, E.; Kuzyakov, Y. Active microorganisms in soil: Critical review of estimation criteria and approaches. Soil Biol. Biochem. 2013, 67, 192–211. [Google Scholar] [CrossRef]
- Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 2005, 3, 470–478. [Google Scholar] [CrossRef]
- Stefani, F.O.P.; Bell, T.H.; Marchand, C.; de la Providencia, I.E.; Yassimi, A.E.; St-Arnaud, M.; Hijri, M. Culture-dependent and -independent methods capture different microbial community fractions in hydrocarbon-contaminated soils. PLoS ONE 2015, 10, e0128272. [Google Scholar] [CrossRef] [Green Version]
- Anguita-Maeso, M.; Olivares-García, C.; Haro, C.; Imperial, J.; Navas-Cortés, J.A.; Landa, B.B. Culture-dependent and culture-independent characterization of the olive xylem microbiota: Effect of sap extraction methods. Front. Plant Sci. 2020, 10, 1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, J.; Albertsen, M. Investigation of detection limits and the influence of DNA extraction and primer choice on the observed microbial communities in drinking water samples using 16S RRNA gene amplicon sequencing. Front. Microbiol. 2018, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Martiny, A.C. High proportions of bacteria are culturable across major biomes. ISME J. 2019, 13, 2125–2128. [Google Scholar] [CrossRef] [PubMed]
- Steen, A.D.; Crits-Christoph, A.; Carini, P.; DeAngelis, K.M.; Fierer, N.; Lloyd, K.G.; Cameron Thrash, J. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 2019, 13, 3126–3130. [Google Scholar] [CrossRef] [Green Version]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, R.; Kahn, D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2004, 2, 621–631. [Google Scholar] [CrossRef]
- Pajares, S.; Bohannan, B.J.M. Ecology of nitrogen fixing, nitrifying, and denitrifying microorganisms in tropical forest soils. Front. Microbiol. 2016, 7, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isobe, K.; Ise, Y.; Kato, H.; Oda, T.; Vincenot, C.E.; Koba, K.; Tateno, R.; Senoo, K.; Ohte, N. Consequences of microbial diversity in forest nitrogen cycling: Diverse ammonifiers and specialized ammonia oxidizers. ISME J. 2020, 14, 12–25. [Google Scholar] [CrossRef] [Green Version]
- Stursová, M.; Zifčáková, L.; Leigh, M.B.; Burgess, R.; Baldrian, P. Cellulose utilization in forest litter and soil: Identification of bacterial and fungal decomposers. FEMS Microbiol. Ecol. 2012, 80, 735–746. [Google Scholar] [CrossRef]
- DeAngelis, K.M.; Allgaier, M.; Chavarria, Y.; Fortney, J.L.; Hugenholtz, P.; Simmons, B.; Sublette, K.; Silver, W.L.; Hazen, T.C. Characterization of trapped lignin-degrading microbes in tropical forest soil. PLoS ONE 2011, 6, e19306. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.E.; Chang, M.C.Y. Exploring bacterial lignin degradation. Curr. Opin. Chem. Biol. 2014, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabugao, K.G.; Timm, C.M.; Carrell, A.A.; Childs, J.; Lu, T.-Y.S.; Pelletier, D.A.; Weston, D.J.; Norby, R.J. Root and rhizosphere bacterial phosphatase activity varies with tree species and soil phosphorus availability in Puerto Rico tropical forest. Front. Plant Sci. 2017, 8, 1834. [Google Scholar] [CrossRef] [Green Version]
- Hayat, R.; Ali, S.; Amara, U.; Khalid, R.; Ahmed, I. Soil beneficial bacteria and their role in plant growth promotion: A review. Ann. Microbiol. 2010, 60, 579–598. [Google Scholar] [CrossRef]
- Lennon, J.T.; Jones, S.E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 2011, 9, 119–130. [Google Scholar] [CrossRef]
- Chen, Q.-L.; Ding, J.; Zhu, D.; Hu, H.-W.; Delgado-Baquerizo, M.; Ma, Y.-B.; He, J.-Z.; Zhu, Y.-G. Rare microbial taxa as the major drivers of ecosystem multifunctionality in long-term fertilized soils. Soil Biol. Biochem. 2020, 141, 107686. [Google Scholar] [CrossRef]
- Pedrós-Alió, C. The rare bacterial biosphere. Annu. Rev. Mar. Sci. 2011, 4, 449–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.D.J.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 2015, 13, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Hogan, C.S.; Klimowicz, A.K.; Linske, M.; McManus, P.S.; Handelsman, J. Culturing captures members of the soil rare biosphere. Environ. Microbiol. 2012, 14, 2247–2252. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 2013, 41, e1. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Sansupa, C.; Wahdan, S.F.M.; Hossen, S.; Disayathanoowat, T.; Wubet, T.; Purahong, W. Can we use functional annotation of prokaryotic taxa (FAPROTAX) to assign the ecological functions of soil bacteria? Appl. Sci. 2021, 11, 688. [Google Scholar] [CrossRef]
- Oksanen, F.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.4-4. 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 10 March 2021).
- Team, R. A language and environment for statistical computing. Computing 2006, 1. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and 574 data analysis. Palaeontol. Electron. 2001, 4, art.4. [Google Scholar]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Wnuk, E.; Waśko, A.; Walkiewicz, A.; Bartmiński, P.; Bejger, R.; Mielnik, L.; Bieganowski, A. The effects of humic substances on DNA isolation from soils. PeerJ 2020, 8, e9378. [Google Scholar] [CrossRef]
- Xie, K.; Deng, Y.; Zhang, X.; Wang, X.; Kang, G.; Bai, L.; Huang, H. Biases in prokaryotic community amplicon sequencing affected by DNA extraction methods in both saline and non-saline soil. Front. Microbiol. 2018, 9, 1796. [Google Scholar] [CrossRef]
- Sansupa, C.; Purahong, W.; Wubet, T.; Tiansawat, P.; Pathom-Aree, W.; Teaumroong, N.; Chantawannakul, P.; Buscot, F.; Elliott, S.; Disayathanoowat, T. Soil bacterial communities and their associated functions for forest restoration on a limestone mine in northern Thailand. PLoS ONE 2021, 16, e0248806. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Niu, S.; Liu, X.; Wang, J. Short-term response of the soil bacterial community to differing wildfire severity in Pinus tabulaeformis stands. Sci. Rep. 2019, 9, 1148. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Penttinen, P.; Mikkonen, A.; Stoddard, F.L.; Lindström, K. Response of soil bacterial community diversity and composition to time, fertilization, and plant species in a sub-boreal climate. Front. Microbiol. 2020, 11, 1780. [Google Scholar] [CrossRef]
- Yan, D.; Bissett, A.; Gellie, N.; Mills, J.G.; Lowe, A.J.; Breed, M.F. Soil bacterial community differences along a coastal restoration chronosequence. Plant Ecol. 2020, 221, 795–811. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil PH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheuer, F.; Nacke, H.; Schöning, I.; Schrumpf, M.; Daniel, R. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci. Rep. 2016, 6, 33696. [Google Scholar] [CrossRef] [Green Version]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef]
- Purahong, W.; Krüger, D. A better understanding of functional roles of fungi in the decomposition process: Using precursor RRNA containing ITS regions as a marker for the active fungal community. Ann. For. Sci. 2012, 69, 659–662. [Google Scholar] [CrossRef]
- Whang, K.; Hattori, T. Oligotrophic bacteria from rendzina forest soil. Antonie van Leeuwenhoek 1988, 54, 19–36. [Google Scholar] [CrossRef]
- Campanharo, J.C.; Kielak, A.M.; Castellane, T.C.L.; Kuramae, E.E.; de Macedo Lemos, E.G. Optimized medium culture for Acidobacteria subdivision 1 strains. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.; Di Lonardo, D.P.; Bodelier, P.L.E. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol. Ecol. 2017, 93, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Li, Q.; Chen, X.; Jiang, C. Isolation and Cultivation Methods of Actinobacteria; IntechOpen: London, UK, 2016; ISBN 978-953-51-2248-7. [Google Scholar]
- Zengler, K.; Toledo, G.; Rappé, M.; Elkins, J.; Mathur, E.J.; Short, J.M.; Keller, M. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA 2002, 99, 15681–15686. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.J.; Hugenholtz, P.; Sangwan, P.; Osborne, C.A.; Janssen, P.H. Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl. Environ. Microbiol. 2003, 69, 7210–7215. [Google Scholar] [CrossRef] [Green Version]
- Janssen, P.H.; Yates, P.S.; Grinton, B.E.; Taylor, P.M.; Sait, M. Improved culturability of soil bacteria and isolation in pure culture of novel members of the divisions Acidobacteria, Actinobacteria, Proteobacteria, and Verrucomicrobia. Appl. Environ. Microbiol. 2002, 68, 2391–2396. [Google Scholar] [CrossRef] [Green Version]
- Da Rocha, U.N.; Andreote, F.D.; de Azevedo, J.L.; van Elsas, J.D.; van Overbeek, L.S. Cultivation of hitherto-uncultured bacteria belonging to the Verrucomicrobia subdivision 1 from the potato (Solanum tuberosum L.) rhizosphere. J. Soils Sediments 2010, 10, 326–339. [Google Scholar] [CrossRef] [Green Version]
- Ardley, J. Legumes of the thar desert and their nitrogen fixing ensifer symbionts. Plant Soil 2017, 410, 517–520. [Google Scholar] [CrossRef]
- Songsumanus, A.; Tanasupawat, S.; Thawai, C.; Suwanborirux, K.; Kudo, T. Micromonospora humi Sp. nov., isolated from peat swamp forest soil. Int. J. Syst. Evol. Microbiol. 2011, 61, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Palmer, K.; Horn, M.A. Actinobacterial nitrate reducers and proteobacterial denitrifiers are abundant in N2O-metabolizing palsa peat. Appl. Environ. Microbiol. 2012, 78, 5584–5596. [Google Scholar] [CrossRef] [Green Version]
- Barboza, N.R.; Amorim, S.S.; Santos, P.A.; Reis, F.D.; Cordeiro, M.M.; Guerra-Sá, R.; Leão, V.A. Indirect manganese removal by Stenotrophomonas sp. and Lysinibacillus sp. isolated from Brazilian mine water. BioMed Res. Int. 2015, 2015, e925972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangaraj, K.; Kapley, A.; Purohit, H.J. Characterization of diverse acinetobacter isolates for utilization of multiple aromatic compounds. Bioresour. Technol. 2008, 99, 2488–2494. [Google Scholar] [CrossRef]
- Hobel, C.F.V.; Marteinsson, V.T.; Hauksdóttir, S.; Fridjónsson, Ó.H.; Skírnisdóttir, S.; Hreggvidsson, G.Ó.; Kristjánsson, J.K. Use of low nutrient enrichments to access novel amylase genes in silent diversity of thermophiles. World J. Microbiol. Biotechnol. 2004, 20, 801–809. [Google Scholar] [CrossRef]
- Kaminsky, R.; Morales, S.E. Conditionally rare taxa contribute but do not account for changes in soil prokaryotic community structure. Front. Microbiol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetzer, I.; Johst, K.; Schäwe, R.; Banitz, T.; Harms, H.; Chatzinotas, A. The extent of functional redundancy changes as species’ roles shift in different environments. Proc. Natl. Acad. Sci. USA 2015, 112, 14888–14893. [Google Scholar] [CrossRef] [Green Version]
- Trap, J.; Bonkowski, M.; Plassard, C.; Villenave, C.; Blanchart, E. Ecological importance of soil bacterivores for ecosystem functions. Plant Soil 2016, 398, 1–24. [Google Scholar] [CrossRef]
- Fakruddin, M.; Mannan, K.S.B.; Andrews, S. Viable but nonculturable bacteria: Food safety and public health perspective. ISRN Microbiol. 2013, 2013, e703813. [Google Scholar] [CrossRef] [PubMed]
- Shakya, M.; Lo, C.-C.; Chain, P.S.G. Advances and challenges in metatranscriptomic analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Wahdan, S.F.M.; Heintz-Buschart, A.; Sansupa, C.; Tanunchai, B.; Wu, Y.-T.; Schädler, M.; Noll, M.; Purahong, W.; Buscot, F. Targeting the active rhizosphere microbiome of trifolium pratense in grassland evidences a stronger-than-expected belowground biodiversity-ecosystem functioning link. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sansupa, C.; Fareed Mohamed Wahdan, S.; Disayathanoowat, T.; Purahong, W. Identifying Hidden Viable Bacterial Taxa in Tropical Forest Soils Using Amplicon Sequencing of Enrichment Cultures. Biology 2021, 10, 569. https://doi.org/10.3390/biology10070569
Sansupa C, Fareed Mohamed Wahdan S, Disayathanoowat T, Purahong W. Identifying Hidden Viable Bacterial Taxa in Tropical Forest Soils Using Amplicon Sequencing of Enrichment Cultures. Biology. 2021; 10(7):569. https://doi.org/10.3390/biology10070569
Chicago/Turabian StyleSansupa, Chakriya, Sara Fareed Mohamed Wahdan, Terd Disayathanoowat, and Witoon Purahong. 2021. "Identifying Hidden Viable Bacterial Taxa in Tropical Forest Soils Using Amplicon Sequencing of Enrichment Cultures" Biology 10, no. 7: 569. https://doi.org/10.3390/biology10070569
APA StyleSansupa, C., Fareed Mohamed Wahdan, S., Disayathanoowat, T., & Purahong, W. (2021). Identifying Hidden Viable Bacterial Taxa in Tropical Forest Soils Using Amplicon Sequencing of Enrichment Cultures. Biology, 10(7), 569. https://doi.org/10.3390/biology10070569