
Histone H3K4 Methyltransferases as Targets for Drug-Resistant Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
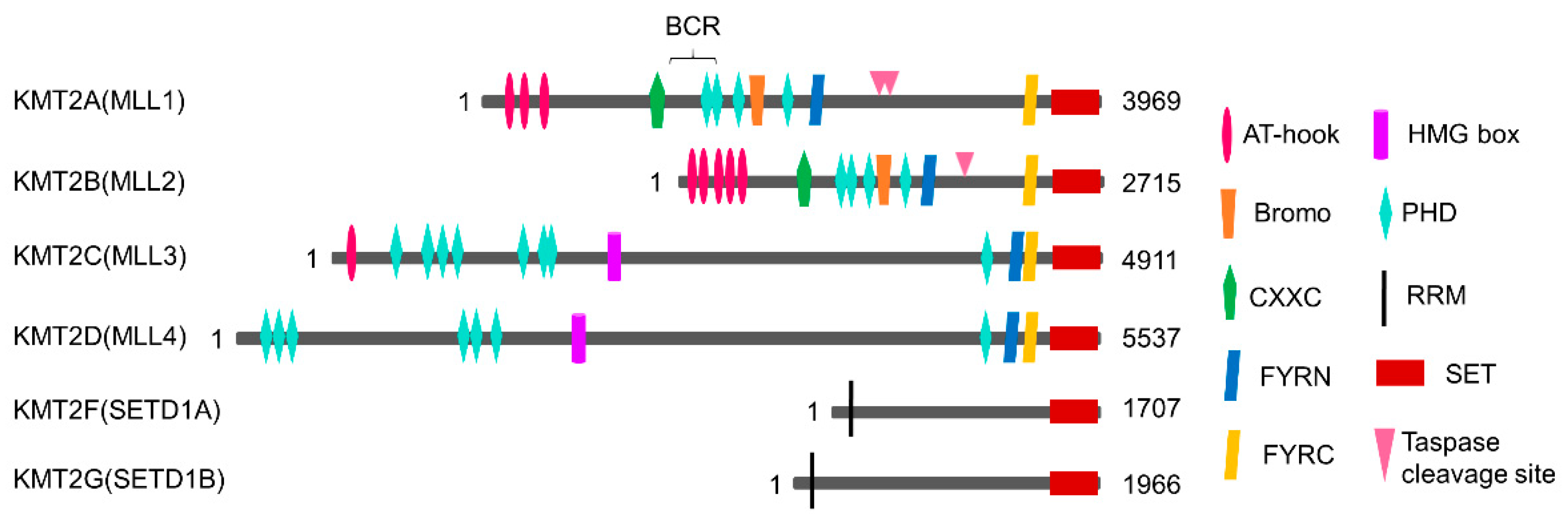
2. Classification of H3K4 Methyltransferases
2.1. KMT2/MLL Family
2.2. SMYD Family
2.3. Other H3K4 Methyltransferases
2.4. Distribution of H3K4 Methylation and H3K4 Readers
3. H3K4 Methyltransferases in Drug-Resistant Cancers
3.1. H3K4 Methyltransferase in Breast Cancer
3.2. H3K4 Methyltransferase in Colorectal Cancer
3.3. H3K4 Methyltransferase in Prostate Cancer
3.4. H3K4 Methyltransferase in Leukemia
3.5. H3K4 Methyltransferase in Gastric Cancer
3.6. H3K4 Methyltransferase in Other Cancers
4. Inhibitors Targeting H3K4-Specific HMTs for Anticancer Therapy
4.1. WDR5 Inhibitors

{kind=link}
| Inhibitor | Structure | Mode of Action | Kd or Ki | Methylation IC50 | PPI IC50 | GI50 | Cancer Cell Type | In Vivo Validation | Ref |
|---|---|---|---|---|---|---|---|---|---|
| MM-102 | 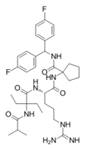 | WDR5–MLL | <1 nM | 400 nM | 2.4 nM | 25 μM | MLL1-rearranged leukemia | [200] | |
| MM-401 |  | WDR5–MLL | <1 nM | 320 nM | 0.9 nM | 5.9~12.6 μM | MLL1-rearranged leukemia | [201] | |
| MM-589 | 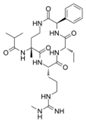 | WDR5–MLL | <1 nM | 12.7 nM | 0.9 nM | 0.21~0.25 μM | MLL1-rearranged leukemia | [202] | |
| WDR5-0103 |  | WDR5–MLL | 450 nM | 39 μM | [203] | ||||
| OICR-9429 | 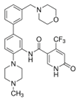 | WDR5–MLL | 30 nM | ~5 μM | C/EBPα-mutant AML Ovarian cancer (with topotecan) Prostate cancer (with cisplatin) | Yes Yes | [204,207] [208] [209] | ||
| Piribedil | 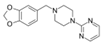 | WDR5–MLL | 180 nM | 65~92 μM | MLL1-rearranged AML | Yes | [205] | ||
| Win6mer |  | WDR5–MLLWDR5–SETD1A | 2.9 nM | 2.2 nM2.5 nM | [206] | ||||
| Compound C6 |  | WDR5–MLL | 0.1 nM | 20 nM | 2.5~6.4 μM | MLL1-rearranged leukemia | [210] | ||
| Compound 16 | 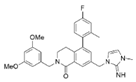 | WDR5–MLL | <0.02 nM | 2.2 nM | 38~78 nM 0.26~0.49 μM | MLL1-rearranged leukemia Neuroblastoma and Burkitt’s lymphoma | [211] | ||
| MCP-1 | 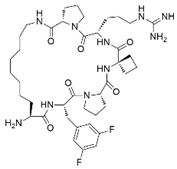 | Menin–MLL | 4.7 nM | 18.5 nM | MLL1-rearranged leukemia | [212] | |||
| MI-2 | 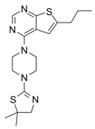 | Menin–MLL | 158 nM | 446 nM | 7.2~18 μM | MLL1-rearranged leukemia | [213] | ||
| MI-2-2 | 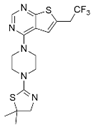 | Menin–MLL | 22 nM | 46 nM | 3 μM | MLL1-rearranged leukemia (with DOT1L inhibitor) | Yes | [214,215] | |
| MI-463 | 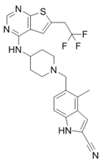 | Menin–MLL | 9.9 nM | 15.3 nM | 0.23 μM | MLL1-rearranged leukemia | Yes | [216] | |
| MI-503 | 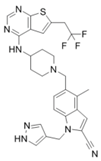 | Menin–MLL | 9.3 nM | 14.7 nM | 0.22 μM 1.5~11.7 μM | MLL1-rearranged leukemia Castration-resistant prostate cancer | Yes Yes | [216] [135] | |
| MI-538 | 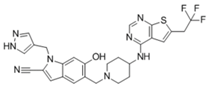 | Menin–MLL | 6.5 nM | 21 nM | 83 nM | MLL1-rearranged leukemia | Yes | [217] | |
| MI-3454 | 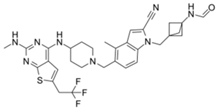 | Menin–MLL | 0.51 nM | 7.6~27.1 nM | MLL1-rearranged or NPM1-mutated leukemia | Yes | [218] | ||
| BAY-155 | 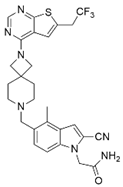 | Menin–MLL | 75 nM | 8 nM | 90~140 nM | MLL1-rearranged leukemia | Yes | [219] | |
| VTP50469 |  | Menin–MLL | 104 pM | 13~37 nM | MLL1-rearranged leukemia | Yes | [220] | ||
| M-89 | 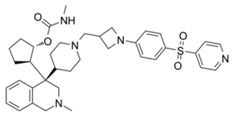 | Menin–MLL | 1.4 nM | 5 nM | 25~55 nM | MLL1-rearranged leukemia | [221] | ||
| M-525 | 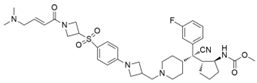 | Menin–MLL | 3.3 nM | 2.3~10.3 nM | The first irreversible Menin inhibitor | [222] | |||
| M-808 | 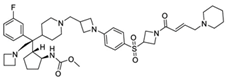 | Menin–MLL | 2.6 nM | 1~4 nM | MLL1-rearranged leukemia | Yes | [223] | ||
| AZ505 | 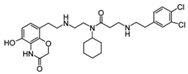 | SMYD2 | 500 nM | 120 nM | MDR-clear cell renal cell carcinoma (ccRCC) Triple-negative breast cancer | Yes | [224] [117] | ||
| LLY-507 | 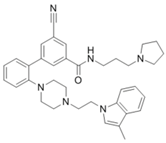 | SMYD2 | <15 nM | 1.5~6 μM 1.77~2.9 μM | Esophageal, liver, and breast cancer cells High-grade serous ovarian carcinomas (HGSOCs). | [225] [226] | |||
| A-893. | 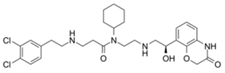 | SMYD2 | 2.8 nM | Lung cancer | [227] | ||||
| BAY598 |  | SMYD2 | 1.1~1.2 nM | 27 nM | <10 μM | Esophageal cancer (combi w/doxorubicin) | Yes | [228] | |
| EPZ031686 |  | SMYD3 | 1.3~4.7 nM | 3 nM | [229] | ||||
| EPZ028862 |  | SMYD3 | 1.8 nM | >40 μM | Esophageal squamous cell carcinoma | [230] | |||
| GSK2807 |  | SMYD3 | 14 nM | 130 nM | [231] | ||||
| Compound 29 | 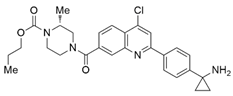 | SMYD3 | 440 nM | 11.7 nM | 17.7 μM (2D) 1.04 μM (3D) | Hepatocarcinoma | [232] | ||
| (R)-PFI-2 | 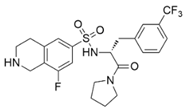 | SETD7/9 | 0.33 nM | 2 nM | Breast cancer cells | [233] |
4.2. Menin Inhibitors
4.3. SMYD Inhibitors
4.4. Other Inhibitors
4.5. Protein Degraders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Mazzio, E.A.; Soliman, K.F. Basic concepts of epigenetics: Impact of environmental signals on gene expression. Epigenetics 2012, 7, 119–130. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Bowman, G.D.; Poirier, M.G. Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 2015, 115, 2274–2295. [Google Scholar] [CrossRef] [Green Version]
- McCabe, M.T.; Mohammad, H.P.; Barbash, O.; Kruger, R.G. Targeting histone methylation in cancer. Cancer J. 2017, 23, 292–301. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, J.; Ma, Y.; Wu, C.; Cui, W.; Wang, L. Histone methyltransferase and drug resistance in cancers. J. Exp. Clin. Cancer Res. 2020, 39, 173. [Google Scholar] [CrossRef]
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal Transduct. Target. Ther. 2020, 5, 143. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. Adv. Genet. 2010, 70, 57–85. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Mirsky, A.E. Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science 1964, 144, 559. [Google Scholar] [CrossRef]
- Murray, K. The occurrence of iε-N-methyl lysine in histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef]
- Schurter, B.T.; Koh, S.S.; Chen, D.; Bunick, G.J.; Harp, J.M.; Hanson, B.L.; Henschen-Edman, A.; Mackay, D.R.; Stallcup, M.R.; Aswad, D.W. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 2001, 40, 5747–5756. [Google Scholar] [CrossRef]
- Guccione, E.; Bassi, C.; Casadio, F.; Martinato, F.; Cesaroni, M.; Schuchlautz, H.; Luscher, B.; Amati, B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007, 449, 933–937. [Google Scholar] [CrossRef]
- Hyllus, D.; Stein, C.; Schnabel, K.; Schiltz, E.; Imhof, A.; Dou, Y.; Hsieh, J.; Bauer, U.M. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007, 21, 3369–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iberg, A.N.; Espejo, A.; Cheng, D.; Kim, D.; Michaud-Levesque, J.; Richard, S.; Bedford, M.T. Arginine methylation of the histone H3 tail impedes effector binding. J. Biol. Chem. 2008, 283, 3006–3010. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huang, Z.Q.; Xia, L.; Feng, Q.; Erdjument-Bromage, H.; Strahl, B.D.; Briggs, S.D.; Allis, C.D.; Wong, J.; Tempst, P.; et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 2001, 293, 853–857. [Google Scholar] [CrossRef]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [Green Version]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Mohan, M.; Herz, H.M.; Shilatifard, A. SnapShot: Histone lysine methylase complexes. Cell 2012, 149, 498–498.e1. [Google Scholar] [CrossRef] [Green Version]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, S.; Schotta, G.; Sorensen, C.S. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013, 41, 2797–2806. [Google Scholar] [CrossRef]
- Morera, L.; Lübbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.; So, C. Epigenetic therapies by targeting aberrant histone methylome in AML: Molecular mechanisms, current preclinical and clinical development. Oncogene 2017, 36, 1753–1759. [Google Scholar] [CrossRef]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilatifard, A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 2008, 20, 341–348. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.R.; Burnett, R.C.; Gill, H.J.; Thirman, M.J.; Mbangkollo, D.; Kipiniak, M.; van Melle, E.; Ziemin-van der Poel, S.; Rowley, J.D.; Diaz, M.O. Cloning of cDNAs of the MLL gene that detect DNA rearrangements and altered RNA transcripts in human leukemic cells with 11q23 translocations. Proc. Natl. Acad. Sci. USA 1992, 89, 11794–11798. [Google Scholar] [CrossRef] [Green Version]
- Tkachuk, D.C.; Kohler, S.; Cleary, M.L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992, 71, 691–700. [Google Scholar] [CrossRef]
- Zeisig, B.B.; Milne, T.; García-Cuéllar, M.P.; Schreiner, S.; Martin, M.E.; Fuchs, U.; Borkhardt, A.; Chanda, S.K.; Walker, J.; Soden, R.; et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol. Cell. Biol. 2004, 24, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Takahashi, Y.H.; Westfield, G.H.; Oleskie, A.N.; Trievel, R.C.; Shilatifard, A.; Skiniotis, G. Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc. Natl. Acad. Sci. USA 2011, 108, 20526–20531. [Google Scholar] [CrossRef] [Green Version]
- Southall, S.M.; Wong, P.S.; Odho, Z.; Roe, S.M.; Wilson, J.R. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol. Cell 2009, 33, 181–191. [Google Scholar] [CrossRef]
- Patel, A.; Dharmarajan, V.; Cosgrove, M.S. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem. 2008, 283, 32158–32161. [Google Scholar] [CrossRef] [Green Version]
- Shinsky, S.A.; Hu, M.; Vought, V.E.; Ng, S.B.; Bamshad, M.J.; Shendure, J.; Cosgrove, M.S. A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J. Mol. Biol. 2014, 426, 2283–2299. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Chen, Y.; Cierpicki, T.; Liu, Y.; Basrur, V.; Lei, M.; Dou, Y. An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS ONE 2010, 5, e14102. [Google Scholar] [CrossRef]
- Van Nuland, R.; Smits, A.H.; Pallaki, P.; Jansen, P.W.; Vermeulen, M.; Timmers, H.M. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol. Cell. Biol. 2013, 33, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, R.; Zhou, M.-M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Ayton, P.M.; Chen, E.H.; Cleary, M.L. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol. Cell. Biol. 2004, 24, 10470–10478. [Google Scholar] [CrossRef] [Green Version]
- Schlichter, A.; Cairns, B.R. Histone trimethylation by Set1 is coordinated by the RRM, autoinhibitory, and catalytic domains. EMBO J. 2005, 24, 1222–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crump, N.T.; Milne, T.A. Why are so many MLL lysine methyltransferases required for normal mammalian development? Cell. Mol. Life Sci. 2019, 76, 2885–2898. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem. 2008, 283, 32162–32175. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Lee, S.Y.; Zhou, B.; Nguyen, U.T.; Muir, T.W.; Tan, S.; Dou, Y. ASH2L regulates ubiquitylation signaling to MLL: Trans-regulation of H3 K4 methylation in higher eukaryotes. Mol. Cell 2013, 49, 1108–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.-M.; Smith, E.R.; Shilatifard, A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 2013, 33, 4745–4754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinsky, S.A.; Monteith, K.E.; Viggiano, S.; Cosgrove, M.S. Biochemical reconstitution and phylogenetic comparison of human SET1 family core complexes involved in histone methylation. J. Biol. Chem. 2015, 290, 6361–6375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Lin, C.; Smith, E.R.; Guo, H.; Sanderson, B.W.; Wu, M.; Gogol, M.; Alexander, T.; Seidel, C.; Wiedemann, L.M.; et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009, 29, 6074–6085. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Garruss, A.S.; Gao, X.; Morgan, M.A.; Cook, M.; Smith, E.R.; Shilatifard, A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1093–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Wang, C.; Xu, S.; Cho, Y.W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2013, 2, e01503. [Google Scholar] [CrossRef]
- Hallson, G.; Hollebakken, R.E.; Li, T.; Syrzycka, M.; Kim, I.; Cotsworth, S.; Fitzpatrick, K.A.; Sinclair, D.A.; Honda, B.M. dSet1 is the main H3K4 di- and tri-methyltransferase throughout Drosophila development. Genetics 2012, 190, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Wang, P.F.; Lee, J.S.; Martin-Brown, S.; Florens, L.; Washburn, M.; Shilatifard, A. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 2008, 28, 7337–7344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Guenther, M.G.; Jenner, R.G.; Chevalier, B.; Nakamura, T.; Croce, C.M.; Canaani, E.; Young, R.A. Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. USA 2005, 102, 8603–8608. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Li, Y.; Liang, S.; Cui, K.; Salz, T.; Yang, H.; Tang, Z.; Gallagher, P.G.; Qiu, Y.; Roeder, R. USF1 and hSET1A mediated epigenetic modifications regulate lineage differentiation and HoxB4 transcription. PLoS Genet. 2013, 9, e1003524. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 2004, 13, 587–597. [Google Scholar] [CrossRef]
- Murai, M.J.; Pollock, J.; He, S.; Miao, H.; Purohit, T.; Yokom, A.; Hess, J.L.; Muntean, A.G.; Grembecka, J.; Cierpicki, T. The same site on the integrase-binding domain of lens epithelium–derived growth factor is a therapeutic target for MLL leukemia and HIV. Blood J. Am. Soc. Hematol. 2014, 124, 3730–3737. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.-W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M. PTIP associates with MLL3-and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef] [Green Version]
- Sirinupong, N.; Brunzelle, J.; Ye, J.; Pirzada, A.; Nico, L.; Yang, Z. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture. J. Biol. Chem. 2010, 285, 40635–40644. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Lambert, J.-P.; Al-Madhoun, A.S.; Elisma, F.; Skerjanc, I.S.; Figeys, D. The tale of two domains: Proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell. Proteom. 2008, 7, 560–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef]
- Spellmon, N.; Holcomb, J.; Trescott, L.; Sirinupong, N.; Yang, Z. Structure and function of SET and MYND domain-containing proteins. Int. J. Mol. Sci. 2015, 16, 1406–1428. [Google Scholar] [CrossRef] [Green Version]
- Foreman, K.W.; Brown, M.; Park, F.; Emtage, S.; Harriss, J.; Das, C.; Zhu, L.; Crew, A.; Arnold, L.; Shaaban, S. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS ONE 2011, 6, e22290. [Google Scholar] [CrossRef]
- Gottlieb, P.D.; Pierce, S.A.; Sims, R.J.; Yamagishi, H.; Weihe, E.K.; Harriss, J.V.; Maika, S.D.; Kuziel, W.A.; King, H.L.; Olson, E.N. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 2002, 31, 25–32. [Google Scholar] [CrossRef]
- Sims, R.J., 3rd; Weihe, E.K.; Zhu, L.; O’Malley, S.; Harriss, J.V.; Gottlieb, P.D. m-Bop, a repressor protein essential for cardiogenesis, interacts with skNAC, a heart- and muscle-specific transcription factor. J. Biol. Chem. 2002, 277, 26524–26529. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Rotllant, J.; Li, H.; DeDeyne, P.; Du, S.J. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl. Acad. Sci. USA 2006, 103, 2713–2718. [Google Scholar] [CrossRef] [Green Version]
- Berkholz, J.; Orgeur, M.; Stricker, S.; Munz, B. skNAC and Smyd1 in transcriptional control. Exp. Cell Res. 2015, 336, 182–191. [Google Scholar] [CrossRef]
- Brown, M.A.; Sims, R.J.; Gottlieb, P.D.; Tucker, P.W. Identification and characterization of Smyd2: A split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 2006, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef]
- Wu, J.; Cheung, T.; Grande, C.; Ferguson, A.D.; Zhu, X.; Theriault, K.; Code, E.; Birr, C.; Keen, N.; Chen, H. Biochemical characterization of human SET and MYND domain-containing protein 2 methyltransferase. Biochemistry 2011, 50, 6488–6497. [Google Scholar] [CrossRef] [PubMed]
- Saddic, L.A.; West, L.E.; Aslanian, A.; Yates, J.R.; Rubin, S.M.; Gozani, O.; Sage, J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Aller, G.S.; Reynoird, N.; Barbash, O.; Huddleston, M.; Liu, S.; Zmoos, A.-F.; McDevitt, P.; Sinnamon, R.; Le, B.; Mas, G. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 2012, 7, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunizaki, M.; Hamamoto, R.; Silva, F.P.; Yamaguchi, K.; Nagayasu, T.; Shibuya, M.; Nakamura, Y.; Furukawa, Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007, 67, 10759–10765. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 2001, 8, 1207–1217. [Google Scholar] [CrossRef]
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, G.M.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656. [Google Scholar] [CrossRef] [Green Version]
- Nishioka, K.; Chuikov, S.; Sarma, K.; Erdjument-Bromage, H.; Allis, C.D.; Tempst, P.; Reinberg, D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002, 16, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Kurash, J.K.; Lei, H.; Shen, Q.; Marston, W.L.; Granda, B.W.; Fan, H.; Wall, D.; Li, E.; Gaudet, F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 2008, 29, 392–400. [Google Scholar] [CrossRef]
- Estève, P.-O.; Chin, H.G.; Benner, J.; Feehery, G.R.; Samaranayake, M.; Horwitz, G.A.; Jacobsen, S.E.; Pradhan, S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 5076–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontaki, H.; Talianidis, I. Lysine methylation regulates E2F1-induced cell death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z.; Xu, C.; Leng, X.; Cao, H.; Ouyang, G.; Xiao, W. Repression of hypoxia-inducible factor α signaling by Set7-mediated methylation. Nucleic Acids Res. 2015, 43, 5081–5098. [Google Scholar] [CrossRef] [Green Version]
- Oudhoff, M.J.; Braam, M.J.; Freeman, S.A.; Wong, D.; Rattray, D.G.; Wang, J.; Antignano, F.; Snyder, K.; Refaeli, I.; Hughes, M.R. SETD7 controls intestinal regeneration and tumorigenesis by regulating Wnt/β-catenin and Hippo/YAP signaling. Dev. Cell 2016, 37, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Yoshida, K.; Matsui, Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 2005, 438, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Eram, M.S.; Bustos, S.P.; Lima-Fernandes, E.; Siarheyeva, A.; Senisterra, G.; Hajian, T.; Chau, I.; Duan, S.; Wu, H.; Dombrovski, L.; et al. Trimethylation of histone H3 lysine 36 by human methyltransferase PRDM9 protein. J. Biol. Chem. 2014, 289, 12177–12188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, N.R.; Parvanov, E.D.; Baker, C.L.; Walker, M.; Petkov, P.M.; Paigen, K. The Meiotic Recombination Activator PRDM9 Trimethylates Both H3K36 and H3K4 at Recombination Hotspots In Vivo. PLoS Genet. 2016, 12, e1006146. [Google Scholar] [CrossRef]
- Wu, H.; Mathioudakis, N.; Diagouraga, B.; Dong, A.; Dombrovski, L.; Baudat, F.; Cusack, S.; de Massy, B.; Kadlec, J. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep. 2013, 5, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Kim, T.; Buratowski, S. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell 2009, 137, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 2006, 442, 86–90. [Google Scholar] [CrossRef]
- Lauberth, S.M.; Nakayama, T.; Wu, X.; Ferris, A.L.; Tang, Z.; Hughes, S.H.; Roeder, R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 2013, 152, 1021–1036. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Fan, X.; Sha, Q.-Q.; Wang, H.-H.; Li, B.-T.; Dai, X.-X.; Shen, L.; Liu, J.; Wang, L.; Liu, K. CFP1 regulates histone H3K4 trimethylation and developmental potential in mouse oocytes. Cell Rep. 2017, 20, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Li, J.; Song, T.; Lu, M.; Kan, P.-Y.; Lee, M.G.; Sha, B.; Shi, X. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 2010, 285, 9322–9326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chen, Z.; Mao, Z.; Zhang, H.; Ding, X.; Chen, S.; Zhang, X.; Xu, R.; Zhu, B. Nucleolar protein Spindlin1 recognizes H3K4 methylation and stimulates the expression of rRNA genes. EMBO Rep. 2011, 12, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. NUP98-PHF23 is a chromatin modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Cantù, C.; Valenta, T.; Hausmann, G.; Vilain, N.; Aguet, M.; Basler, K. The Pygo2-H3K4me2/3 interaction is dispensable for mouse development and Wnt signaling-dependent transcription. Development 2013, 140, 2377–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, R.J., III; Chen, C.-F.; Santos-Rosa, H.; Kouzarides, T.; Patel, S.S.; Reinberg, D. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J. Biol. Chem. 2005, 280, 41789–41792. [Google Scholar] [CrossRef] [Green Version]
- Jeong, K.W.; Kim, K.; Situ, A.J.; Ulmer, T.S.; An, W.; Stallcup, M.R. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol. 2011, 18, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Bian, C.; Xu, C.; Ruan, J.; Lee, K.K.; Burke, T.L.; Tempel, W.; Barsyte, D.; Li, J.; Wu, M.; Zhou, B.O. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 2011, 30, 2829–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, T.; Binda, O.; Champagne, K.S.; Kuo, A.J.; Johnson, K.; Chang, H.Y.; Simon, M.D.; Kutateladze, T.G.; Gozani, O. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol. Cell 2009, 33, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Wilting, R.H.; Dannenberg, J.-H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updates 2012, 15, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Mo, R.; Rao, S.M.; Zhu, Y.-J. Identification of the MLL2 complex as a coactivator for estrogen receptor α. J. Biol. Chem. 2006, 281, 15714–15720. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, T.G.; Kallakury, B.V.; Sheehan, C.E.; Bartlett, M.B.; Ganesan, N.; Preet, A.; Ross, J.S.; FitzGerald, K.T. Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon. Cancer Cell Int. 2010, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gala, K.; Li, Q.; Sinha, A.; Razavi, P.; Dorso, M.; Sanchez-Vega, F.; Chung, Y.R.; Hendrickson, R.; Hsieh, J.J.; Berger, M. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERα enhancer function. Oncogene 2018, 37, 4692–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-S.; Lee, M.-H.; Lee, M.-O. Histone methyltransferases regulate the transcriptional expression of ERα and the proliferation of tamoxifen-resistant breast cancer cells. Breast Cancer Res. Treat. 2020, 180, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Sharma, A.; Dhar, S.S.; Lee, S.-H.; Gu, B.; Chan, C.-H.; Lin, H.-K.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salz, T.; Deng, C.; Pampo, C.; Siemann, D.; Qiu, Y.; Brown, K.; Huang, S. Histone methyltransferase hSETD1A is a novel regulator of metastasis in breast cancer. Mol. Cancer Res. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, K.; Matsuda, S.; Yae, T.; Drapkin, B.J.; Morris, R.; Boukhali, M.; Niederhoffer, K.; Comaills, V.; Dubash, T.; Nieman, L. SETD1A protects from senescence through regulation of the mitotic gene expression program. Nat. Commun. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Tang, Z.; Chen, C.W.; Shimada, M.; Koche, R.P.; Wang, L.H.; Nakadai, T.; Chramiec, A.; Krivtsov, A.V.; Armstrong, S.A.; et al. A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol. Cell 2017, 67, 308–321.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Collings, C.K.; Zhao, Z.; Cozzolino, K.A.; Ma, Q.; Liang, K.; Marshall, S.A.; Sze, C.C.; Hashizume, R.; Savas, J.N.; et al. A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolism. Genes Dev. 2017, 31, 2056–2066. [Google Scholar] [CrossRef]
- Li, L.X.; Zhou, J.X.; Calvet, J.P.; Godwin, A.K.; Jensen, R.A.; Li, X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018, 9, 326. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.L.; Kim, Y.W.; Jin, H.L.; Kang, H.; Lee, E.K.; Stallcup, M.R.; Jeong, K.W. Aberrant expression of SETD1A promotes survival and migration of estrogen receptor α-positive breast cancer cells. Int. J. Cancer 2018, 143, 2871–2883. [Google Scholar] [CrossRef] [Green Version]
- Fenizia, C.; Bottino, C.; Corbetta, S.; Fittipaldi, R.; Floris, P.; Gaudenzi, G.; Carra, S.; Cotelli, F.; Vitale, G.; Caretti, G. SMYD3 promotes the epithelial–mesenchymal transition in breast cancer. Nucleic Acids Res. 2019, 47, 1278–1293. [Google Scholar] [CrossRef] [Green Version]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, M.L.; Wang, C.; Dong, Q.Q.; Miao, Z.; Chen, X.Y.; Wang, N.; He, H.P.; Zhang, T.C.; Luo, X.G. SET and MYND domain-containing protein 3 inhibits tumor cell sensitivity to cisplatin. Oncol. Lett. 2020, 19, 3469–3476. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Zhou, J.; Zhao, Y.; Zheng, J.; Cui, L. SET7/9 promotes multiple malignant processes in breast cancer development via RUNX2 activation and is negatively regulated by TRIM21. Cell Death Dis. 2020, 11, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Li, X.; Yu, Y.; Ma, L.; Liu, S.; Zong, X.; Zheng, Q. SETD7 is a prognosis predicting factor of breast cancer and regulates redox homeostasis. Oncotarget 2017, 8, 94080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, K.; Jia, D.; Kapoor-Vazirani, P.; Powell, D.R.; Collins, R.E.; Sharma, D.; Peng, J.; Cheng, X.; Vertino, P.M. Regulation of estrogen receptor α by the SET7 lysine methyltransferase. Mol. Cell 2008, 30, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, M.; Sánchez-Del-Campo, L.; González-Guerrero, R.; Martínez-Barba, E.; Piñero-Madrona, A.; Cabezas-Herrera, J.; Rodríguez-López, J. Tumor suppressor SET9 guides the epigenetic plasticity of breast cancer cells and serves as an early-stage biomarker for predicting metastasis. Oncogene 2016, 35, 6143–6152. [Google Scholar] [CrossRef]
- Larsson, C.; Cordeddu, L.; Siggens, L.; Pandzic, T.; Kundu, S.; He, L.; Ali, M.A.; Pristovšek, N.; Hartman, K.; Ekwall, K. Restoration of KMT2C/MLL3 in human colorectal cancer cells reinforces genome-wide H3K4me1 profiles and influences cell growth and gene expression. Clin. Epigenet. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.H.; Lee, S.; Yang, Q.H.; Lee, D.K.; Lee, S.K.; Roeder, R.G.; Lee, J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. USA 2009, 106, 8513–8518. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-L.; Du, X.; Tan, L.-N.; Deng, F.-H.; Zhou, B.-Y.; Zhou, H.-J.; Zhu, H.-Y.; Chu, Y.; Liu, D.-L.; Tan, Y.-Y. SET7 interacts with HDAC6 and suppresses the development of colon cancer through inactivation of HDAC6. Am. J. Transl. Res. 2020, 12, 602. [Google Scholar]
- Rahnamoun, H.; Hong, J.; Sun, Z.; Lee, J.; Lu, H.; Lauberth, S.M. Mutant p53 regulates enhancer-associated H3K4 monomethylation through interactions with the methyltransferase MLL4. J. Biol. Chem. 2018, 293, 13234–13246. [Google Scholar] [CrossRef] [Green Version]
- Salz, T.; Li, G.; Kaye, F.; Zhou, L.; Qiu, Y.; Huang, S. hSETD1A regulates Wnt target genes and controls tumor growth of colorectal cancer cells. Cancer Res. 2014, 74, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Teng, H.; Wang, Y.; Liao, G.; Weng, L.; Li, Y.; Wang, X.; Jin, J.; Jiao, C.; Chen, L.; et al. SET1A-Mediated Mono-Methylation at K342 Regulates YAP Activation by Blocking Its Nuclear Export and Promotes Tumorigenesis. Cancer Cell 2018, 34, 103–118.e9. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Oh, H.R.; Choi, M.R.; Gwak, M.; An, C.H.; Chung, Y.J.; Yoo, N.J.; Lee, S.H. Frameshift mutation of a histone methylation-related gene SETD1B and its regional heterogeneity in gastric and colorectal cancers with high microsatellite instability. Hum. Pathol. 2014, 45, 1674–1681. [Google Scholar] [CrossRef]
- Ren, H.; Wang, Z.; Chen, Y.; Liu, Y.; Zhang, S.; Zhang, T.; Li, Y. SMYD2-OE promotes oxaliplatin resistance in colon cancer through MDR1/P-glycoprotein via MEK/ERK/AP1 pathway. OncoTargets Ther. 2019, 12, 2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, R.; Khan, A.P.; Asangani, I.A.; Cieslik, M.; Prensner, J.R.; Wang, X.; Iyer, M.K.; Jiang, X.; Borkin, D.; Escara-Wilke, J.; et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat. Med. 2015, 21, 344–352. [Google Scholar] [CrossRef]
- Lv, S.; Ji, L.; Chen, B.; Liu, S.; Lei, C.; Liu, X.; Qi, X.; Wang, Y.; Leung, E.L.-H.; Wang, H. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene 2018, 37, 1354–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.; Wen, H.; Shan, X.; Li, J.; Wu, Y.; Yu, X.; Huang, W.; Wei, Q. Loss of KMT2D induces prostate cancer ROS-mediated DNA damage by suppressing the enhancer activity and DNA binding of antioxidant transcription factor FOXO3. Epigenetics 2019, 14, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Vieira, F.Q.; Costa-Pinheiro, P.; Almeida-Rios, D.; Graça, I.; Monteiro-Reis, S.; Simões-Sousa, S.; Carneiro, I.; Sousa, E.J.; Godinho, M.I.; Baltazar, F. SMYD3 contributes to a more aggressive phenotype of prostate cancer and targets Cyclin D2 through H4K20me3. Oncotarget 2015, 6, 13644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Wang, C.; Wang, K.; Liu, L.; Shen, Q.; Yan, K.; Sun, X.; Chen, J.; Liu, J.; Ren, H. SMYD3 as an oncogenic driver in prostate cancer by stimulation of androgen receptor transcription. J. Natl. Cancer Inst. 2013, 105, 1719–1728. [Google Scholar] [CrossRef] [Green Version]
- Gaughan, L.; Stockley, J.; Wang, N.; McCracken, S.R.; Treumann, A.; Armstrong, K.; Shaheen, F.; Watt, K.; McEwan, I.J.; Wang, C. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011, 39, 1266–1279. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sargsyan, D.; Zhang, C.; Wu, R.; Yang, Y.; Kong, A.-N. Transcriptomic Analysis of Histone Methyltransferase Setd7 Knockdown and Phenethyl Isothiocyanate in Human Prostate Cancer Cells. Anticancer Res. 2018, 38, 6069–6083. [Google Scholar] [CrossRef] [Green Version]
- Sowalsky, A.G.; Xia, Z.; Wang, L.; Zhao, H.; Chen, S.; Bubley, G.J.; Balk, S.P.; Li, W. Whole transcriptome sequencing reveals extensive unspliced mRNA in metastatic castration-resistant prostate cancer. Mol. Cancer Res. 2015, 13, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jin, M.; Park, S.J.; Seo, S.-Y.; Jeong, K.W. SETD1A promotes proliferation of castration-resistant Prostate cancer cells via FOXM1 transcription. Cancers 2020, 12, 1736. [Google Scholar] [CrossRef]
- Djabali, M.; Selleri, L.; Parry, P.; Bower, M.; Young, B.D.; Evans, G.A. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 1992, 2, 113–118. [Google Scholar] [CrossRef]
- Ziemin-van der Poel, S.; McCabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; LeBeau, M.M.; Rowley, J.D. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, M.; Lin, C.; Guest, E.; Shilatifard, A. Licensed to elongate: A molecular mechanism for MLL-based leukaemogenesis. Nat. Rev. Cancer 2010, 10, 721–728. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011, 25, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivtsov, A.V.; Feng, Z.; Lemieux, M.E.; Faber, J.; Vempati, S.; Sinha, A.U.; Xia, X.; Jesneck, J.; Bracken, A.P.; Silverman, L.B.; et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Thiel, A.T.; Blessington, P.; Zou, T.; Feather, D.; Wu, X.; Yan, J.; Zhang, H.; Liu, Z.; Ernst, P.; Koretzky, G.A.; et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 2010, 17, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshii, T.; Cifani, P.; Feng, Z.; Huang, C.-H.; Koche, R.; Chen, C.-W.; Delaney, C.D.; Lowe, S.W.; Kentsis, A.; Armstrong, S.A. A non-catalytic function of SETD1A regulates cyclin K and the DNA damage response. Cell 2018, 172, 1007–1021.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Anastassiadis, K.; Kranz, A.; Stewart, A.F.; Arndt, K.; Waskow, C.; Yokoyama, A.; Jones, K.; Neff, T.; Lee, Y. MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer Cell 2017, 31, 755–770.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabello, D.D.A.; Ferreira, V.; Berzoti-Coelho, M.G.; Burin, S.M.; Magro, C.L.; Cacemiro, M.D.C.; Simões, B.P.; Saldanha-Araujo, F.; de Castro, F.A.; Pittella-Silva, F. MLL2/KMT2D and MLL3/KMT2C expression correlates with disease progression and response to imatinib mesylate in chronic myeloid leukemia. Cancer Cell Int. 2018, 18, 26. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, L.H.T.; de Andrade, R.V.; Felipe, M.S.S.; Motoyama, A.B.; Silva, F.P. SMYD2 is highly expressed in pediatric acute lymphoblastic leukemia and constitutes a bad prognostic factor. Leuk. Res. 2014, 38, 496–502. [Google Scholar] [CrossRef]
- Zipin-Roitman, A.; Aqaqe, N.; Yassin, M.; Biechonski, S.; Amar, M.; van Delft, M.F.; Gan, O.I.; McDermott, S.P.; Buzina, A.; Ketela, T.; et al. SMYD2 lysine methyltransferase regulates leukemia cell growth and regeneration after genotoxic stress. Oncotarget 2017, 8, 16712–16727. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Y.; Wang, X.; Gao, L.; Yu, W.; Dong, W.F. Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells. J. Cancer 2017, 8, 2069–2078. [Google Scholar] [CrossRef] [Green Version]
- Houle, A.A.; Gibling, H.; Lamaze, F.C.; Edgington, H.A.; Soave, D.; Fave, M.-J.; Agbessi, M.; Bruat, V.; Stein, L.D.; Awadalla, P. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. 2018, 28, 1611–1620. [Google Scholar] [CrossRef] [Green Version]
- Hussin, J.; Sinnett, D.; Casals, F.; Idaghdour, Y.; Bruat, V.; Saillour, V.; Healy, J.; Grenier, J.-C.; De Malliard, T.; Busche, S. Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res. 2013, 23, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Deng, Z.; Tang, Y.; Deng, Z.; Li, M. Downregulation of KMT2D suppresses proliferation and induces apoptosis of gastric cancer. Biochem. Biophys. Res. Commun. 2018, 504, 129–136. [Google Scholar] [CrossRef]
- Cho, S.-J.; Yoon, C.; Lee, J.H.; Chang, K.K.; Lin, J.-X.; Kim, Y.-H.; Kook, M.-C.; Aksoy, B.A.; Park, D.J.; Ashktorab, H. KMT2C mutations in diffuse-type gastric adenocarcinoma promote epithelial-to-mesenchymal transition. Clin. Cancer Res. 2018, 24, 6556–6569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chai, H.; Xu, X.; Yu, J.; Gu, Y. Histone methyltransferase SETD1A interacts with HIF1α to enhance glycolysis and promote cancer progression in gastric cancer. Mol. Oncol. 2020, 14, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Ichikawa, D.; Hirajima, S.; Nagata, H.; Nishimura, Y.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Konishi, H. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br. J. Cancer 2015, 112, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, Q.-T.; Liu, Y.-P.; Dong, Q.-Q.; Hu, H.-J.; Miao, Z.; Li, S.; Liu, Y.; Zhou, H.; Zhang, T.-C. ATM signaling pathway is implicated in the SMYD3-mediated proliferation and migration of gastric cancer cells. J. Gastric Cancer 2017, 17, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Xu, Q.; Chen, P.; Yu, C.; Ye, L.; Huang, C.; Li, T. Effect of SMYD3 on biological behavior and H3K4 methylation in bladder cancer. Cancer Manag. Res. 2019, 11, 8125. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, Y.; Koda, Y.; Byeon, S.J.; Shimada, S.; Nishikawaji, T.; Sakamoto, A.; Chen, Y.; Kojima, K.; Kawano, T.; Eishi, Y.; et al. Reduced expression of SET7/9, a histone mono-methyltransferase, is associated with gastric cancer progression. Oncotarget 2016, 7, 3966–3983. [Google Scholar] [CrossRef] [Green Version]
- Stavrovskaya, A.; Stromskaya, T. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochemistry (Moscow) 2008, 73, 592–604. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef]
- Huo, H.; Magro, P.G.; Pietsch, E.C.; Patel, B.B.; Scotto, K.W. Histone methyltransferase MLL1 regulates MDR1 transcription and chemoresistance. Cancer Res. 2010, 70, 8726–8735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Fang, L.; Heuberger, J.; Kranz, A.; Schipper, J.; Scheckenbach, K.; Vidal, R.O.; Sunaga-Franze, D.Y.; Müller, M.; Wulf-Goldenberg, A. The Wnt-driven Mll1 epigenome regulates salivary gland and head and neck cancer. Cell Rep. 2019, 26, 415–428.e5. [Google Scholar] [CrossRef] [Green Version]
- Wend, P.; Fang, L.; Zhu, Q.; Schipper, J.H.; Loddenkemper, C.; Kosel, F.; Brinkmann, V.; Eckert, K.; Hindersin, S.; Holland, J.D.; et al. Wnt/beta-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013, 32, 1977–1989. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J. Natl. Cancer Inst. 2017, 109, djw283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawkins, J.B.; Wang, J.; Maniati, E.; Heward, J.A.; Koniali, L.; Kocher, H.M.; Martin, S.A.; Chelala, C.; Balkwill, F.R.; Fitzgibbon, J. Reduced expression of histone methyltransferases KMT2C and KMT2D correlates with improved outcome in pancreatic ductal adenocarcinoma. Cancer Res. 2016, 76, 4861–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudureheman, A.; Ainiwaer, J.; Hou, Z.; Niyaz, M.; Turghun, A.; Hasim, A.; Zhang, H.; Lu, X.; Sheyhidin, I. High MLL2 expression predicts poor prognosis and promotes tumor progression by inducing EMT in esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augert, A.; Zhang, Q.; Bates, B.; Cui, M.; Wang, X.; Wildey, G.; Dowlati, A.; MacPherson, D. Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J. Thorac. Oncol. 2017, 12, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Rampias, T.; Karagiannis, D.; Avgeris, M.; Polyzos, A.; Kokkalis, A.; Kanaki, Z.; Kousidou, E.; Tzetis, M.; Kanavakis, E.; Stravodimos, K. The lysine-specific methyltransferase KMT 2C/MLL 3 regulates DNA repair components in cancer. EMBO Rep. 2019, 20, e46821. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, G.; Chen, B.; Wang, Y.; Guo, L.; Cao, L.; Ren, C.; Wen, L.; Liao, N. Association between histone lysine methyltransferase KMT2C mutation and clinicopathological factors in breast cancer. Biomed. Pharmacother. 2019, 116, 108997. [Google Scholar] [CrossRef]
- Xiong, W.; Deng, H.; Huang, C.; Zen, C.; Jian, C.; Ye, K.; Zhong, Z.; Zhao, X.; Zhu, L. MLL3 enhances the transcription of PD-L1 and regulates anti-tumor immunity. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 454–463. [Google Scholar] [CrossRef]
- Wu, J.; Chai, H.; Li, F.; Ren, Q.; Gu, Y. SETD1A augments sorafenib primary resistance via activating YAP in hepatocellular carcinoma. Life Sci. 2020, 260, 118406. [Google Scholar] [CrossRef]
- Li, T.; Zheng, Q.; An, J.; Wu, M.; Li, H.; Gui, X.; Pu, H.; Lu, D. SET1A Cooperates With CUDR to Promote Liver Cancer Growth and Hepatocyte-like Stem Cell Malignant Transformation Epigenetically. Mol. Ther. 2016, 24, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Redd, P.S.; Ibrahim, M.L.; Klement, J.D.; Sharman, S.K.; Paschall, A.V.; Yang, D.; Nayak-Kapoor, A.; Liu, K. SETD1B Activates iNOS Expression in Myeloid-Derived Suppressor Cells. Cancer Res. 2017, 77, 2834–2843. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kumari, N.; Rai, A.; Singh, S.K.; Kakkar, N.; Prasad, R. Expression and clinical significance of COMPASS family of histone methyltransferases in clear cell renal cell carcinoma. Gene 2018, 674, 31–36. [Google Scholar] [CrossRef]
- Peveling-Oberhag, J.; Wolters, F.; Döring, C.; Walter, D.; Sellmann, L.; Scholtysik, R.; Lucioni, M.; Schubach, M.; Paulli, M.; Biskup, S. Whole exome sequencing of microdissected splenic marginal zone lymphoma: A study to discover novel tumor-specific mutations. BMC Cancer 2015, 15, 773. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, S.; Imoto, I.; Tsuda, H.; Kozaki, K.-i.; Muramatsu, T.; Shimada, Y.; Aiko, S.; Yoshizumi, Y.; Ichikawa, D.; Otsuji, E. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009, 30, 1139–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, S.-R.; Zuo, X.-C.; He, Y.; Fang, W.-J.; Wang, C.-J.; Zou, H.; Chen, P.; Huang, L.-F.; Huang, L.-H.; Xiang, H. Positive expression of SMYD2 is associated with poor prognosis in patients with primary hepatocellular carcinoma. J. Cancer 2018, 9, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Chen, F.; Fei, X.; Yang, X.; Lu, X. Overexpression of SET and MYND domain-containing protein 2 (SMYD2) is associated with tumor progression and poor prognosis in patients with papillary thyroid carcinoma. Med. Sci. Monit. 2018, 24, 7357. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Wei, M. Inhibition of SMYD2 sensitized cisplatin to resistant cells in NSCLC through activating p53 pathway. Front. Oncol. 2019, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Ding, B.; Liu, H.; Zhang, Y.; Zeng, J.; Hu, J.; Yao, W.; Yu, G.; An, R.; Chen, Z. Inhibition of SMYD2 suppresses tumor progression by down-regulating microRNA-125b and attenuates multi-drug resistance in renal cell carcinoma. Theranostics 2019, 9, 8377. [Google Scholar] [CrossRef]
- Pires-Luís, A.S.; Vieira-Coimbra, M.; Vieira, F.Q.; Costa-Pinheiro, P.; Silva-Santos, R.; Dias, P.C.; Antunes, L.; Lobo, F.; Oliveira, J.; Gonçalves, C.S. Expression of histone methyltransferases as novel biomarkers for renal cell tumor diagnosis and prognostication. Epigenetics 2015, 10, 1033–1043. [Google Scholar] [CrossRef]
- Oliveira-Santos, W.; Rabello, D.A.; Lucena-Araujo, A.R.; de Oliveira, F.M.; Rego, E.M.; Silva, F.P.; Saldanha-Araujo, F. Residual expression of SMYD2 and SMYD3 is associated with the acquisition of complex karyotype in chronic lymphocytic leukemia. Tumor Biol. 2016, 37, 9473–9481. [Google Scholar] [CrossRef]
- Mazur, P.K.; Reynoird, N.; Khatri, P.; Jansen, P.W.; Wilkinson, A.W.; Liu, S.; Barbash, O.; Van Aller, G.S.; Huddleston, M.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef]
- Jiang, Y.; Lyu, T.; Che, X.; Jia, N.; Li, Q.; Feng, W. Overexpression of SMYD3 in ovarian cancer is associated with ovarian cancer proliferation and apoptosis via methylating H3K4 and H4K20. J. Cancer 2019, 10, 4072. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Tsai, C.-H.; Wang, P.-Y.; Teng, S.-C. SMYD3 promotes homologous recombination via regulation of H3K4-mediated gene expression. Sci. Rep. 2017, 7, 3842. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xie, B.-h.; Lin, W.-h.; Huang, Y.-h.; Ni, J.-y.; Hu, J.; Cui, W.; Zhou, J.; Shen, L.; Xu, L.-f. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene 2019, 38, 4948–4961. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, L.; Pan, Y.; Ma, X.; Liu, L.; Wang, W.; You, W. SMYD3 overexpression indicates poor prognosis and promotes cell proliferation, migration and invasion in non-small cell lung cancer. Int. J. Oncol. 2020, 57, 756–766. [Google Scholar] [CrossRef]
- Liu, Q.; Geng, H.; Xue, C.; Beer, T.M.; Qian, D.Z. Functional regulation of hypoxia inducible factor-1α by SET9 lysine methyltransferase. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Wang, D.; Liu, X.; Gu, B.; Du, Y.; Wei, F.Z.; Cao, L.L.; Song, B.; Lu, X.; Yang, Q.; et al. SET7/9 regulates cancer cell proliferation by influencing β-catenin stability. FASEB J. 2015, 29, 4313–4323. [Google Scholar] [CrossRef] [PubMed]
- Chern, T.R.; Liu, L.; Petrunak, E.; Stuckey, J.A.; Wang, M.; Bernard, D.; Zhou, H.; Lee, S.; Dou, Y.; Wang, S. Discovery of Potent Small-Molecule Inhibitors of MLL Methyltransferase. ACS Med. Chem. Lett. 2020, 11, 1348–1352. [Google Scholar] [CrossRef]
- Karatas, H.; Townsend, E.C.; Cao, F.; Chen, Y.; Bernard, D.; Liu, L.; Lei, M.; Dou, Y.; Wang, S. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein–protein interaction. J. Am. Chem. Soc. 2013, 135, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Townsend, E.C.; Karatas, H.; Xu, J.; Li, L.; Lee, S.; Liu, L.; Chen, Y.; Ouillette, P.; Zhu, J. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 2014, 53, 247–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatas, H.; Li, Y.; Liu, L.; Ji, J.; Lee, S.; Chen, Y.; Yang, J.; Huang, L.; Bernard, D.; Xu, J. Discovery of a highly potent, cell-permeable macrocyclic peptidomimetic (MM-589) targeting the WD repeat domain 5 protein (WDR5)—Mixed lineage leukemia (MLL) protein–protein interaction. J. Med. Chem. 2017, 60, 4818–4839. [Google Scholar] [CrossRef] [PubMed]
- Senisterra, G.; Wu, H.; Allali-Hassani, A.; Wasney, G.A.; Barsyte-Lovejoy, D.; Dombrovski, L.; Dong, A.; Nguyen, K.T.; Smil, D.; Bolshan, Y. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 2013, 449, 151–159. [Google Scholar] [CrossRef]
- Grebien, F.; Vedadi, M.; Getlik, M.; Giambruno, R.; Grover, A.; Avellino, R.; Skucha, A.; Vittori, S.; Kuznetsova, E.; Smil, D. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 2015, 11, 571–578. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, X.; Yang, H.; Yan, J.; Fu, X.; Wei, R.; Xu, X.; Zhang, Z.; Yu, A.; Zhou, K. Piribedil disrupts the MLL1-WDR5 interaction and sensitizes MLL-rearranged acute myeloid leukemia (AML) to doxorubicin-induced apoptosis. Cancer Lett. 2018, 431, 150–160. [Google Scholar] [CrossRef]
- Alicea-Velázquez, N.L.; Shinsky, S.A.; Loh, D.M.; Lee, J.-H.; Skalnik, D.G.; Cosgrove, M.S. Targeted disruption of the interaction between WD-40 repeat protein 5 (WDR5) and mixed lineage leukemia (MLL)/SET1 family proteins specifically inhibits MLL1 and SETd1A methyltransferase complexes. J. Biol. Chem. 2016, 291, 22357–22372. [Google Scholar] [CrossRef] [Green Version]
- Getlik, M.; Smil, D.; Zepeda-Velazquez, C.; Bolshan, Y.; Poda, G.; Wu, H.; Dong, A.; Kuznetsova, E.; Marcellus, R.; Senisterra, G.; et al. Structure-Based Optimization of a Small Molecule Antagonist of the Interaction Between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1). J. Med. Chem. 2016, 59, 2478–2496. [Google Scholar] [CrossRef]
- Cao, L.; Wu, G.; Zhu, J.; Tan, Z.; Shi, D.; Wu, X.; Tang, M.; Li, Z.; Hu, Y.; Zhang, S.; et al. Genotoxic stress-triggered β-catenin/JDP2/PRMT5 complex facilitates reestablishing glutathione homeostasis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, X.; He, H.; Peng, S.; Zhang, Y.; Zhang, J.; Cheng, L.; Liu, S.; Huang, M.; Xie, R.; et al. WD repeat domain 5 promotes chemoresistance and Programmed Death-Ligand 1 expression in prostate cancer. Theranostics 2021, 11, 4809–4824. [Google Scholar] [CrossRef]
- Aho, E.R.; Wang, J.; Gogliotti, R.D.; Howard, G.C.; Phan, J.; Acharya, P.; Macdonald, J.D.; Cheng, K.; Lorey, S.L.; Lu, B.; et al. Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019, 26, 2916–2928.e13. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Teuscher, K.B.; Aho, E.R.; Alvarado, J.R.; Mills, J.J.; Meyers, K.M.; Gogliotti, R.D.; Han, C.; Macdonald, J.D.; Sai, J.; et al. Discovery and Structure-Based Optimization of Potent and Selective WD Repeat Domain 5 (WDR5) Inhibitors Containing a Dihydroisoquinolinone Bicyclic Core. J. Med. Chem. 2020, 63, 656–675. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, L.; Huang, J.; Bernard, D.; Karatas, H.; Navarro, A.; Lei, M.; Wang, S. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein–protein interaction. J. Med. Chem. 2013, 56, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Dafflon, C.; Craig, V.J.; Mereau, H.; Grasel, J.; Schacher Engstler, B.; Hoffman, G.; Nigsch, F.; Gaulis, S.; Barys, L.; Ito, M.; et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Murai, M.J.; He, S.; Lund, G.; Hartley, T.; Purohit, T.; Reddy, G.; Chruszcz, M.; Grembecka, J.; Cierpicki, T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [Google Scholar] [CrossRef] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Borkin, D.; Pollock, J.; Kempinska, K.; Purohit, T.; Li, X.; Wen, B.; Zhao, T.; Miao, H.; Shukla, S.; He, M.; et al. Property Focused Structure-Based Optimization of Small Molecule Inhibitors of the Protein-Protein Interaction between Menin and Mixed Lineage Leukemia (MLL). J. Med. Chem. 2016, 59, 892–913. [Google Scholar] [CrossRef] [Green Version]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [Green Version]
- Brzezinka, K.; Nevedomskaya, E.; Lesche, R.; Haegebarth, A.; Ter Laak, A.; Fernández-Montalván, A.E.; Eberspaecher, U.; Werbeck, N.D.; Moenning, U.; Siegel, S. Characterization of the Menin-MLL interaction as therapeutic cancer target. Cancers 2020, 12, 201. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673.e11. [Google Scholar] [CrossRef]
- Aguilar, A.; Zheng, K.; Xu, T.; Xu, S.; Huang, L.; Fernandez-Salas, E.; Liu, L.; Bernard, D.; Harvey, K.P.; Foster, C.; et al. Structure-Based Discovery of M-89 as a Highly Potent Inhibitor of the Menin-Mixed Lineage Leukemia (Menin-MLL) Protein-Protein Interaction. J. Med. Chem. 2019, 62, 6015–6034. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Aguilar, A.; Xu, T.; Zheng, K.; Huang, L.; Stuckey, J.; Chinnaswamy, K.; Bernard, D.; Fernandez-Salas, E.; Liu, L.; et al. Design of the First-in-Class, Highly Potent Irreversible Inhibitor Targeting the Menin-MLL Protein-Protein Interaction. Angew. Chem. 2018, 57, 1601–1605. [Google Scholar] [CrossRef]
- Xu, S.; Aguilar, A.; Huang, L.; Xu, T.; Zheng, K.; McEachern, D.; Przybranowski, S.; Foster, C.; Zawacki, K.; Liu, Z.; et al. Discovery of M-808 as a Highly Potent, Covalent, Small-Molecule Inhibitor of the Menin-MLL Interaction with Strong In Vivo Antitumor Activity. J. Med. Chem. 2020, 63, 4997–5010. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Larsen, N.A.; Howard, T.; Pollard, H.; Green, I.; Grande, C.; Cheung, T.; Garcia-Arenas, R.; Cowen, S.; Wu, J. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Allali-Hassani, A.; Antonysamy, S.; Chang, S.; Chen, L.H.; Curtis, C.; Emtage, S.; Fan, L.; Gheyi, T.; Li, F. LLY-507, a cell-active, potent, and selective inhibitor of protein-lysine methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. [Google Scholar] [CrossRef] [Green Version]
- Kukita, A.; Sone, K.; Oda, K.; Hamamoto, R.; Kaneko, S.; Komatsu, M.; Wada, M.; Honjoh, H.; Kawata, Y.; Kojima, M.; et al. Histone methyltransferase SMYD2 selective inhibitor LLY-507 in combination with poly ADP ribose polymerase inhibitor has therapeutic potential against high-grade serous ovarian carcinomas. Biochem. Biophys. Res. Commun. 2019, 513, 340–346. [Google Scholar] [CrossRef]
- Sweis, R.F.; Wang, Z.; Algire, M.; Arrowsmith, C.H.; Brown, P.J.; Chiang, G.G.; Guo, J.; Jakob, C.G.; Kennedy, S.; Li, F. Discovery of A-893, a new cell-active benzoxazinone inhibitor of lysine methyltransferase SMYD2. ACS Med. Chem. Lett. 2015, 6, 695–700. [Google Scholar] [CrossRef]
- Eggert, E.; Hillig, R.C.; Koehr, S.; Stockigt, D.; Weiske, J.; Barak, N.; Mowat, J.; Brumby, T.; Christ, C.D.; Ter Laak, A.; et al. Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem. 2016, 59, 4578–4600. [Google Scholar] [CrossRef]
- Mitchell, L.H.; Boriack-Sjodin, P.A.; Smith, S.; Thomenius, M.; Rioux, N.; Munchhof, M.; Mills, J.E.; Klaus, C.; Totman, J.; Riera, T.V.; et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Thomenius, M.J.; Totman, J.; Harvey, D.; Mitchell, L.H.; Riera, T.V.; Cosmopoulos, K.; Grassian, A.R.; Klaus, C.; Foley, M.; Admirand, E.A.; et al. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS ONE 2018, 13, e0197372. [Google Scholar] [CrossRef]
- Van Aller, G.S.; Graves, A.P.; Elkins, P.A.; Bonnette, W.G.; McDevitt, P.J.; Zappacosta, F.; Annan, R.S.; Dean, T.W.; Su, D.S.; Carpenter, C.L.; et al. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure 2016, 24, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liew, S.S.; Lin, G.R.; Poulsen, A.; Ang, M.J.Y.; Chia, B.C.S.; Chew, S.Y.; Kwek, Z.P.; Wee, J.L.K.; Ong, E.H.; et al. Discovery of Irreversible Inhibitors Targeting Histone Methyltransferase, SMYD3. ACS Med. Chem. Lett. 2019, 10, 978–984. [Google Scholar] [CrossRef]
- Barsyte-Lovejoy, D.; Li, F.; Oudhoff, M.J.; Tatlock, J.H.; Dong, A.; Zeng, H.; Wu, H.; Freeman, S.A.; Schapira, M.; Senisterra, G.A. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. USA 2014, 111, 12853–12858. [Google Scholar] [CrossRef] [Green Version]
- Tsuge, M.; Hamamoto, R.; Silva, F.P.; Ohnishi, Y.; Chayama, K.; Kamatani, N.; Furukawa, Y.; Nakamura, Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5′ flanking region of SMYD3 is a risk factor for human cancers. Nat. Genet. 2005, 37, 1104–1107. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, X.; Deng, J.; Pan, Y.; Zhang, L.; Liang, H. SMYD3 overexpression was a risk factor in the biological behavior and prognosis of gastric carcinoma. Tumour Biol. 2015, 36, 2685–2694. [Google Scholar] [CrossRef]
- Feichtinger, J.; Aldeailej, I.; Anderson, R.; Almutairi, M.; Almatrafi, A.; Alsiwiehri, N.; Griffiths, K.; Stuart, N.; Wakeman, J.A.; Larcombe, L.; et al. Meta-analysis of clinical data using human meiotic genes identifies a novel cohort of highly restricted cancer-specific marker genes. Oncotarget 2012, 3, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Allali-Hassani, A.; Szewczyk, M.M.; Ivanochko, D.; Organ, S.L.; Bok, J.; Ho, J.S.Y.; Gay, F.P.; Li, F.; Blazer, L.; Eram, M.S. Discovery of a chemical probe for PRDM9. Nat. Commun. 2019, 10, 5759. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
| Enzyme | Cancer Types | Proposed Mechanism |
|---|---|---|
| MLL1 | Chemotherapy resistant MLL leukemia Castration-resistant prostate cancer Anti-PD-L1/PD-1 resistant pancreatic cancer | Increases MDR-1 expression [170] Activates androgen receptor signaling [135] Increases PD-L1 expression [173] |
| MLL3 | Tamoxifen-resistant breast cancer Anti-PD-L1/PD-1 resistant prostate cancer | Increases ERα expression [111] Increases PD-l expression [179] |
| SETD1A | Tamoxifen-resistant breast cancer Tamoxifen-resistant breast cancer Triple-negative breast cancer Castration-resistant prostate cancer Sorafenib resistant hepatocarcinoma | Increases ERα expression [111] Activates ERα signaling and EGFR expression [119] Activates MMP expression [113] Activates FOXM1 signaling via binding with E2F1 [143] Activates Yes-associate protein [180] |
| SETD1B | Triple-negative breast cancer | Regulates adiponectin receptor 1 signaling [116] |
| SMYD2 | Triple-negative breast cancer Oxaliplatin-resistant colon cancer Cisplatin-resistant non-small cell lung cancer Chemotherapy resistant renal cell carcinoma | Activates STAT3 and the p65 [117] Increases MDR-1 expression [134] Inhibits p53 signaling [188] Increases MDR-1 expression [189] |
| SMYD3 | Cisplatin-resistant breast cancer | Increases WNT10B expression and promote the EMT [120,121] |
| SET7/9 | Anti-estrogen-resistant breast cancer Castration-resistant prostate cancer | Controls the stability of E2F1 and DNMT1 [126] Alters chromatin accessibility via frameshift mutation [142] |
| Drug Name | Status | Mechanism | Cancer Type | Administration | ClinicalTrial.gov ID# |
|---|---|---|---|---|---|
| KO-539 | Phase 1/2a | Menin–MLL1 inhibitor | Relapsed or Refractory Acute Myeloid Leukemia | Oral | NCT04067336 |
| JNJ-75276617 | Phase 1 | Menin–MLL1 inhibitor | Acute Leukemias Acute Myeloid Leukemia Acute Lymphoblastic Leukemia | Oral | NCT04811560 |
| SNDX-5613 | Phase 1/2 | Menin–MLL1 inhibitor | Acute lymphoblastic leukemia (ALL) Mixed phenotype acute leukemia (MPAL). Acute Myeloid Leukemia (AML). NPM1c AML. | Oral | NCT04065399 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Jin, M.; Jeong, K.W. Histone H3K4 Methyltransferases as Targets for Drug-Resistant Cancers. Biology 2021, 10, 581. https://doi.org/10.3390/biology10070581
Yang L, Jin M, Jeong KW. Histone H3K4 Methyltransferases as Targets for Drug-Resistant Cancers. Biology. 2021; 10(7):581. https://doi.org/10.3390/biology10070581
Chicago/Turabian StyleYang, Liu, Mingli Jin, and Kwang Won Jeong. 2021. "Histone H3K4 Methyltransferases as Targets for Drug-Resistant Cancers" Biology 10, no. 7: 581. https://doi.org/10.3390/biology10070581