
Key Roles of De-Domestication and Novel Mutation in Origin and Diversification of Global Weedy Rice




Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. PCR Amplification and Sequencing
2.3. SSR and InDel Genotyping
2.4. Data Score and Analyses
3. Results
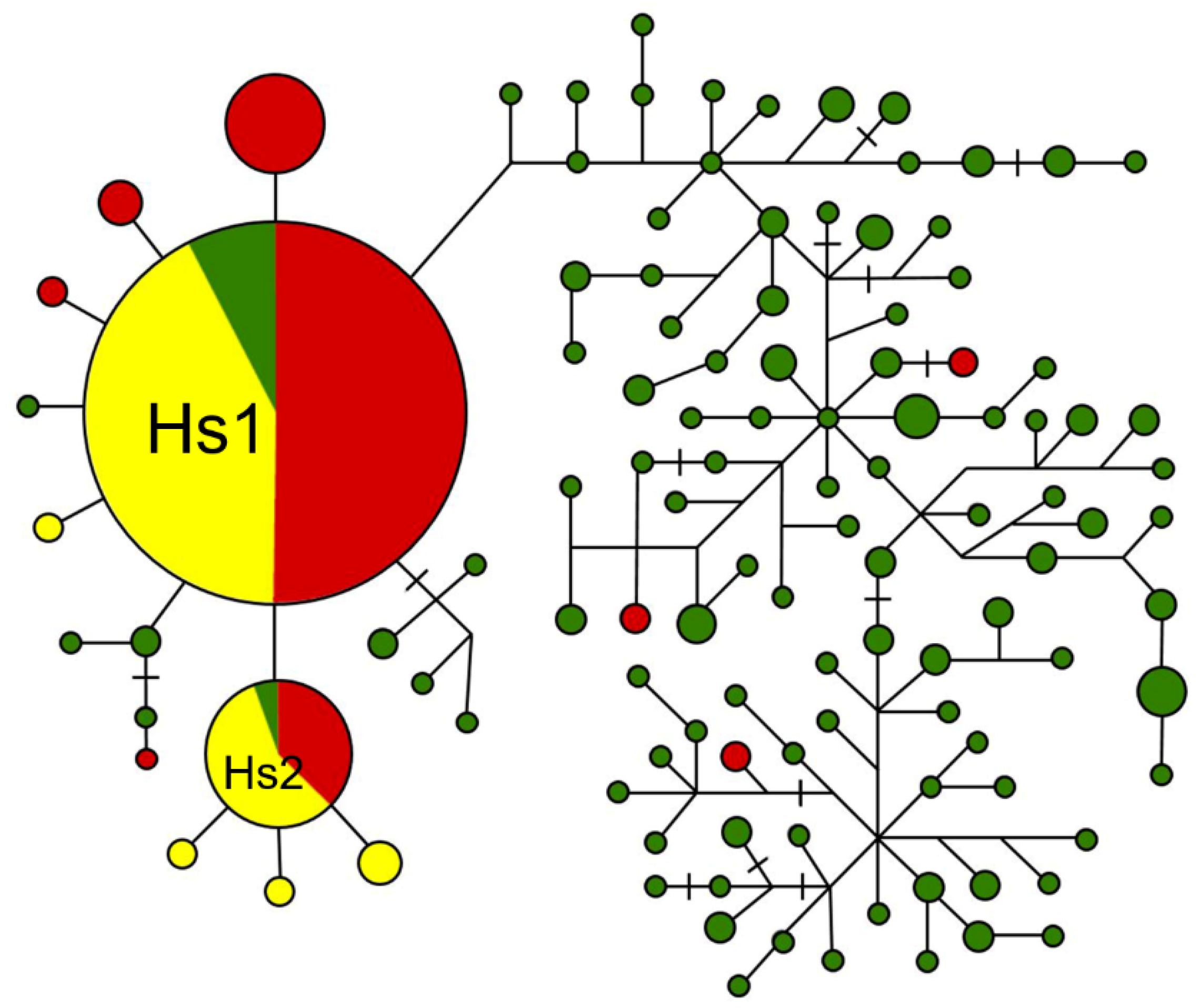
3.1. Sequence Characterization of the sh4 and qsh1 Loci
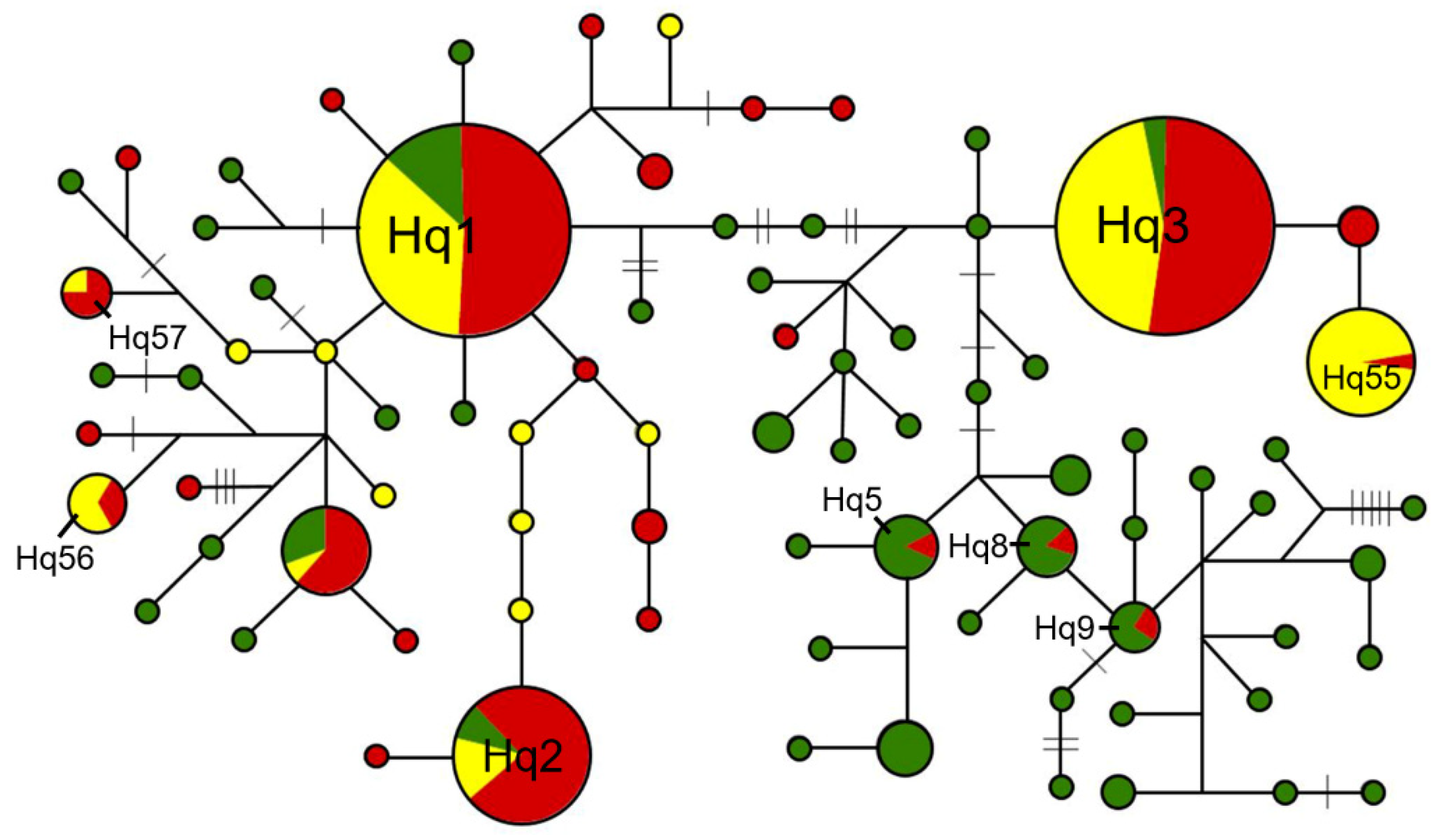
3.2. Haplotype Network Analyses of the sh4 and qsh1 Sequences
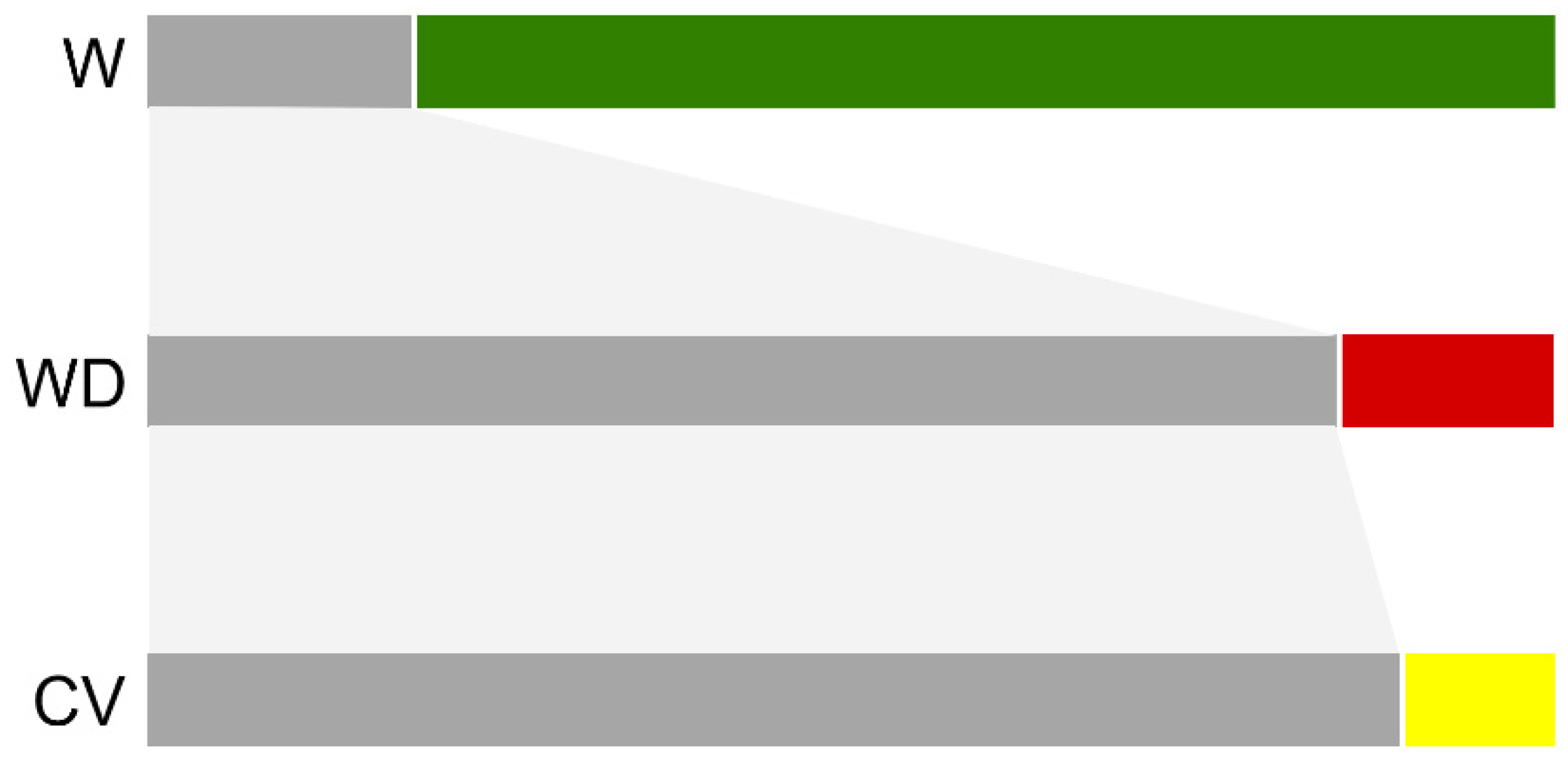
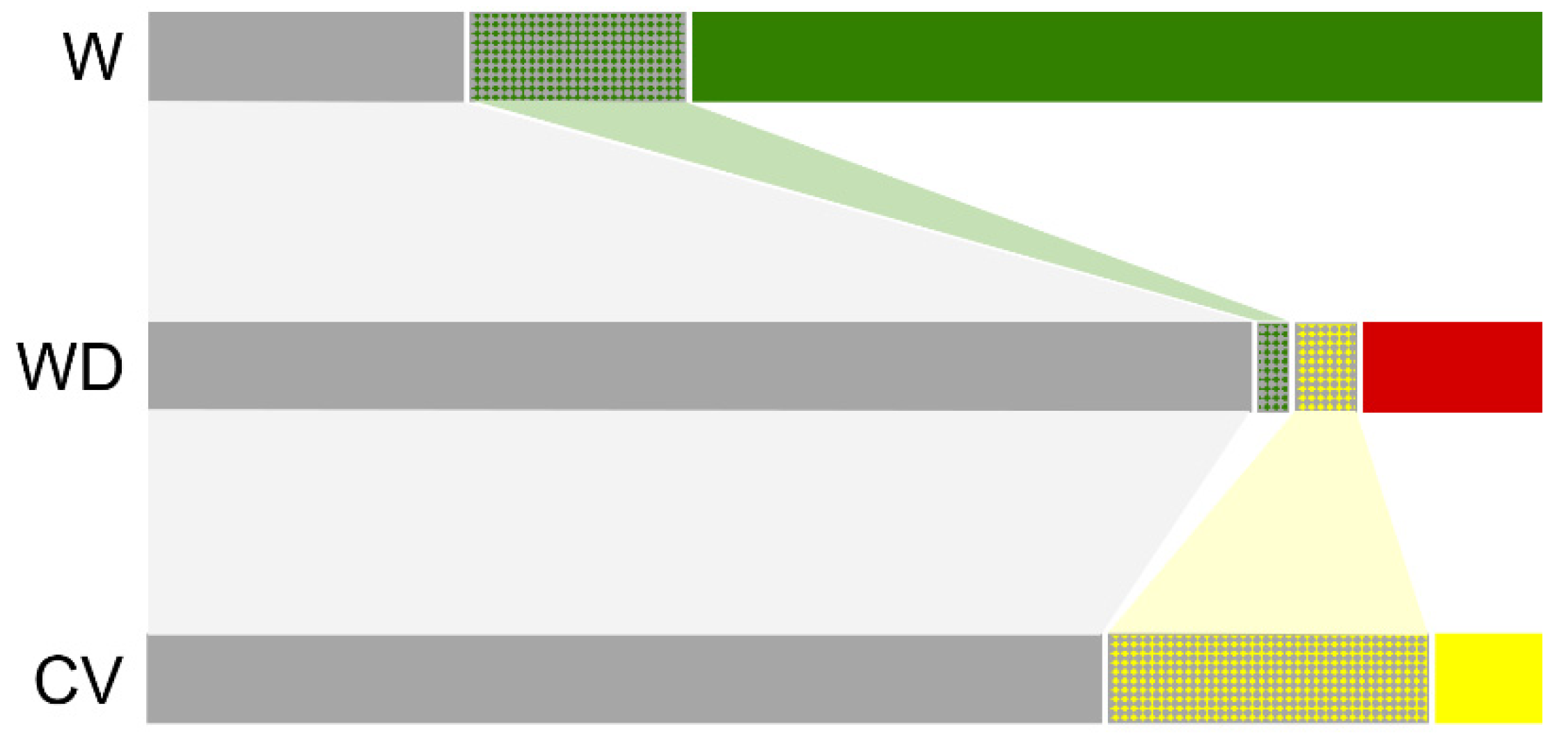
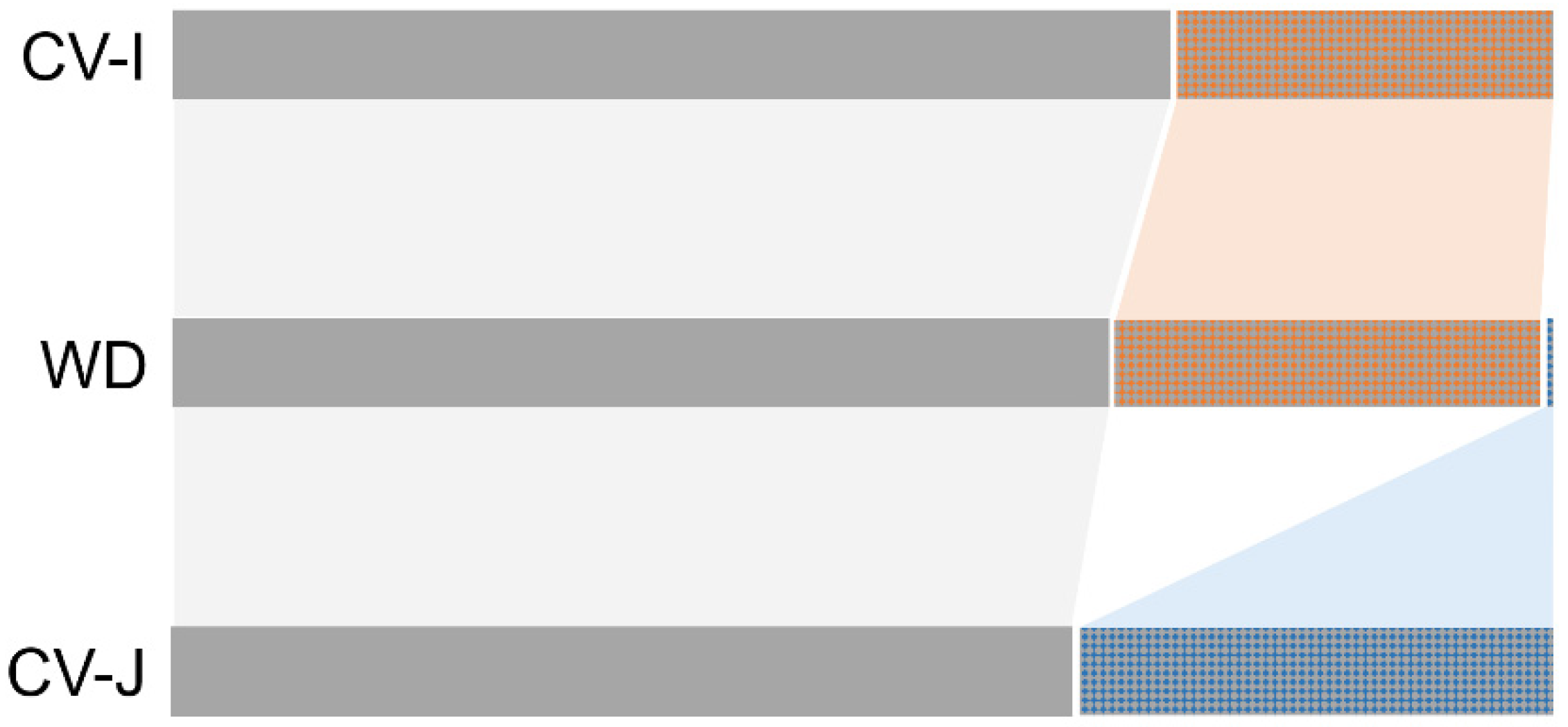
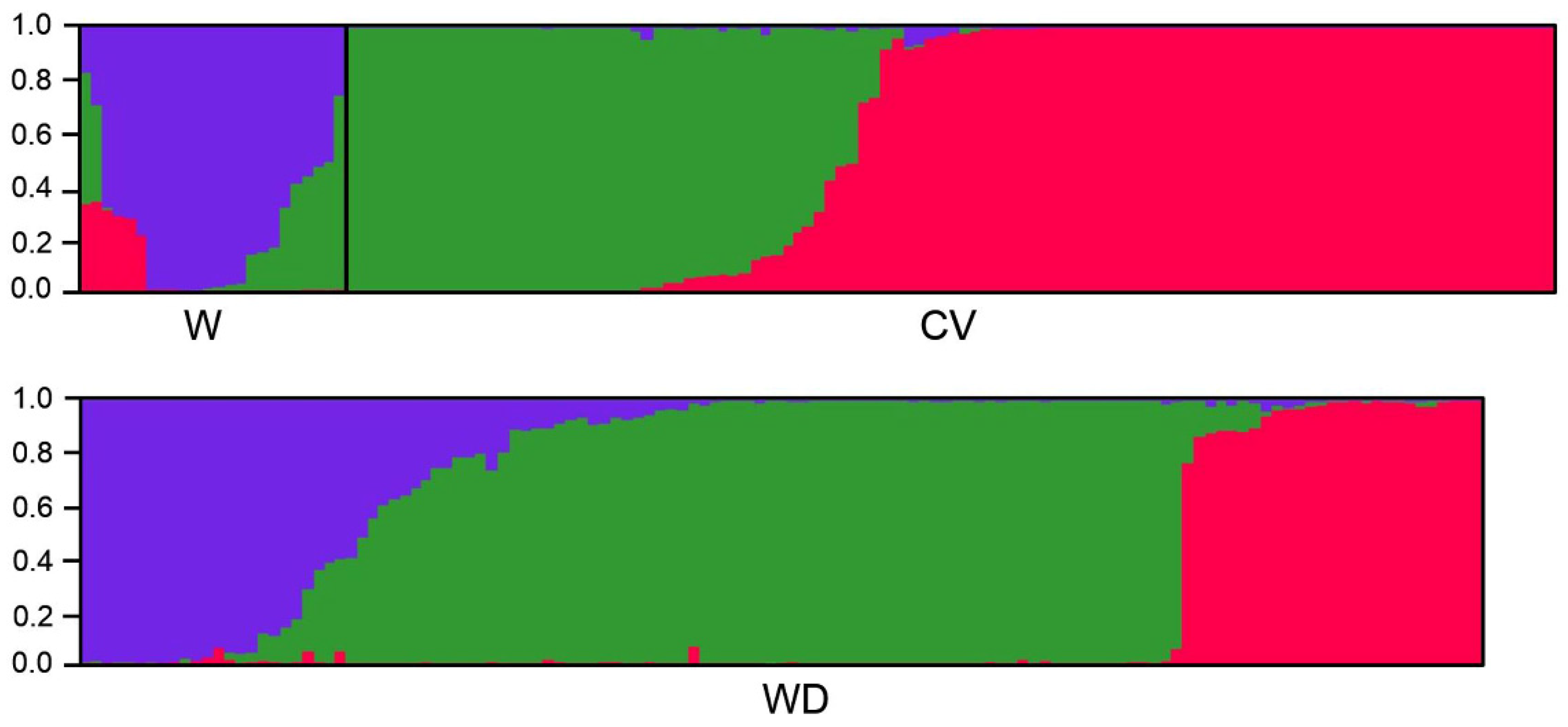
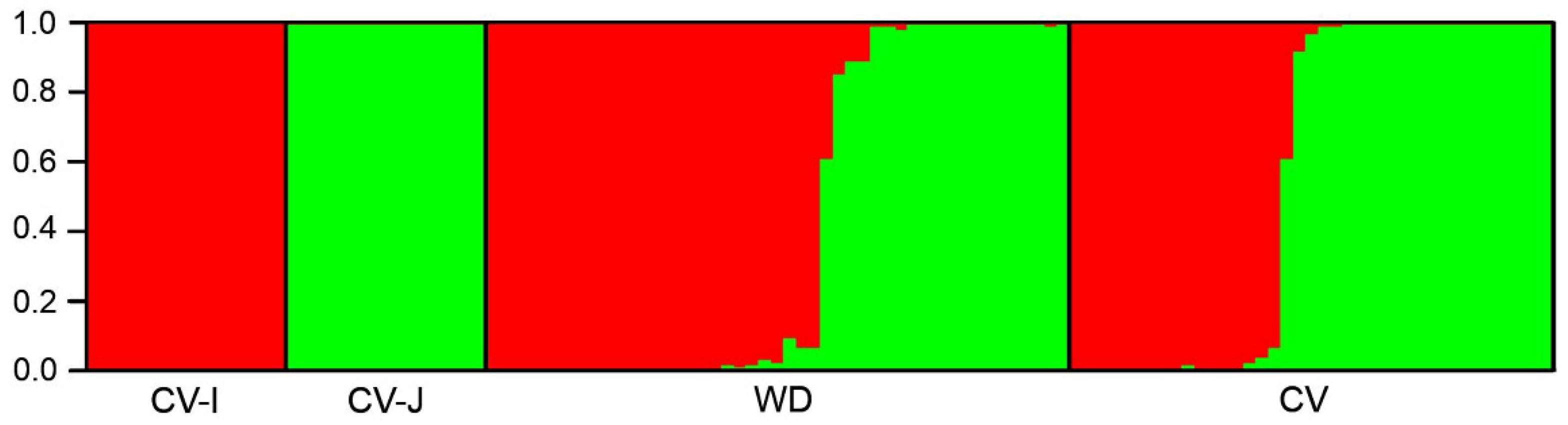
3.3. Genetic Structure of Wild, Cultivated, and Weedy Rice Based on SSR and InDel Molecular Fingerprints
4. Discussion
4.1. De-Domestication Plays the Major Role in the Origin and Evolution of Weedy Rice
4.2. De-Domesticated Weedy Rice Associated with Genetic Divergence of Rice Varieties
4.3. Novel Mutations Promote Further Evolution of Weedy Rice after Its Origins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kwon, S.L.; Smith, R.J.; Talbert, R.E. Comparative growth and development of red rice (Oryza sativa) and rice (O. sativa). Weed Sci. 1992, 40, 57–62. [Google Scholar] [CrossRef]
- Rajcan, I.; Swanton, C. Understanding maize-weed competition: Resource competition, light quality and the whole plant. Field Crop. Res. 2001, 71, 139–150. [Google Scholar] [CrossRef]
- Patracchini, C.; Vidotto, F.; Ferrero, A. Common ragweed (Ambrosia artemisiifolia) growth as affected by plant density and clipping. Weed Technol. 2011, 25, 268–276. [Google Scholar] [CrossRef]
- Oliveira, I.J.D.; Fontes, J.R.A. Weeds as hosts for new crop pests: The case of Protortonia navesi (Hemiptera: Monophlebidae) on cassava in Brazil. Weed Res. 2008, 48, 197–200. [Google Scholar] [CrossRef]
- Chen, G.; Pan, H.P.; Xie, W.; Wang, S.L.; Wu, Q.J.; Fang, Y.; Shi, X.B.; Zhang, Y.J. Virus infection of a weed increases vector attraction to and vector fitness on the weed. Sci. Rep. 2013, 3, 2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delouche, J.C.; Burgos, N.R.; Gealy, D.R.; Zorrilla, D.; Labrada, R.; Larinde, M.; Rosell, C. Weedy rice: Origin, biology, ecology and control. In FAO Plant Production and Protection Paper; FAO: Rome, Italy, 2007. [Google Scholar]
- Nadir, S.; Xiong, H.; Zhu, Q.; Zhang, X.L.; Xu, H.Y.; Li, J.; Dongchen, W.H.; Henry, D.; Guo, X.Q.; Khan, S.; et al. Weedy rice in sustainable rice production. A review. Agron. Sustain. Dev. 2017, 37, 46. [Google Scholar] [CrossRef]
- Food and Agricultural Organization (FAO). The Lurking Menace of Weeds. 2011. Available online: http://www.fao.org/news/story/en/item/29402/icode (accessed on 16 August 2021).
- Green, J.; Barker, J.; Marshall, E.; Froud Williams, R.; Peters, N.; Arnold, G.; Dawson, K.; Karp, A. Microsatellite analysis of the inbreeding grass weed Barren Brome (Anisantha sterilis) reveals genetic diversity at the within- and between-farm scales. Mol. Ecol. 2001, 10, 1035–1045. [Google Scholar] [CrossRef]
- Ishikawa, R.; Toki, N.; Imai, K.; Sato, Y.I.; Yamagishi, H.; Shimamoto, Y.; Ueno, K.; Morishima, H.; Sato, T. Origin of weedy rice grown in Bhutan and the force of genetic diversity. Genet. Resour. Crop Evol. 2005, 52, 395–403. [Google Scholar] [CrossRef]
- Xia, H.B.; Xia, H.; Ellstrand, N.C.; Yang, C.; Lu, B.-R. Rapid evolutionary divergence and ecotypic diversification of germination behavior in weedy rice populations. New Phytol. 2011, 191, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Ma, D.R.; Xu, Z.J.; Deng, H.F.; Chen, W.F.; Yuan, L.P. Utilization of weedy rice for development of japonica hybrid rice (Oryza sativa L.). Plant Sci. 2011, 180, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.S.; Sato, Y.I.; Morishima, H. Genetic characterization of weedy rice (Oryza sativa L.) based on morpho-physiology, isozymes and RAPD markers. Theor. Appl. Genet. 1997, 94, 316–321. [Google Scholar] [CrossRef]
- Westman, A.L.; Kresovich, S. Simple sequence repeat (SSR)-based marker variation in Brassica nigra genebank accessions and weed populations. Euphytica 1999, 109, 85–92. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Prentice, H.C.; Hancock, J.F. Gene flow and introgression from domesticated plants into their wild relatives. Annu. Rev. Ecol. Syst. 1999, 30, 539–563. [Google Scholar] [CrossRef]
- Mispan, M.S.; Zhang, L.H.; Feng, J.H.; Gu, X.Y. Quantitative trait locus and haplotype analyses of wild and crop-mimic traits in U.S. weedy rice. G3 Genes Genomes Genet. 2013, 3, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gressel, J. Crop Ferality and Volunteerism; CRC Press: Los Angeles, CA, USA, 2005. [Google Scholar]
- Li, L.F.; Li, Y.L.; Jia, Y.L.; Caicedo, A.L.; Olsen, K.M. Signatures of adaptation in the weedy rice genome. Nat. Genet. 2017, 49, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Wang, Z.; Song, Z.J.; Wang, L.; Liu, Y.S.; Bao, Y.; Lu, B.-R. Origins of weedy rice revealed by polymorphisms of chloroplast DNA sequences and nuclear microsatellites. J. Syst. Evol. 2021, 59, 316–325. [Google Scholar] [CrossRef]
- Gu, X.Y.; Chen, Z.X.; Foley, M. Inheritance of seed dormancy in weedy rice. Crop Sci. 2003, 43, 835. [Google Scholar] [CrossRef]
- Jiang, X.Q.; Zhu, X.Y.; Lu, B.-R. Induced dormancy in weedy rice seeds by changes of hormone contents under soil burial: Implications in adaptive evolution and weed control. J. Syst. Evol. 2021, e12737. [Google Scholar] [CrossRef]
- Gressel, J.; Valverde, B. A strategy to provide long-term control of weedy rice while mitigating herbicide resistance transgene flow, and its potential use for other crops with related weeds. Pest Manag. Sci. 2009, 65, 723–731. [Google Scholar] [CrossRef]
- Londo, J.; Schaal, B.A. Origins and population genetics of weedy rice in the USA. Mol. Ecol. 2007, 16, 4523–4535. [Google Scholar] [CrossRef]
- Grimm, A.; Fogliatto, S.; Nick, P.; Ferrero, A.; Vidotto, F. Microsatellite markers reveal multiple origins for Italian weedy rice. Ecol. Evol. 2013, 3, 4786–4798. [Google Scholar] [CrossRef]
- Fuller, D.Q.; Qin, L.; Zheng, Y.F.; Zhao, Z.J.; Chen, X.G.; Hosoya, L.; Sun, G.P. The domestication process and domestication rate in rice: Spikelet bases from the Lower Yangtze. Science 2009, 323, 1607–1610. [Google Scholar] [CrossRef] [Green Version]
- Li, C.B.; Zhou, A.L.; Sang, T. Rice domestication by reducing shattering. Science 2006, 311, 1936–1939. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Zhu, J.; Fu, F.; Ye, C.Y.; Wang, W.D.; Mao, L.F.; Lin, Z.X.; Chen, L.; Zhang, H.Q.; Guo, L.B.; et al. Genome re-sequencing suggested a weedy rice origin from domesticated indica-japonica hybridization: A case study from southern China. Planta 2014, 240, 1353–1363. [Google Scholar] [CrossRef]
- Konishi, S.; Izawa, T.; Lin, S.; Ebana, K.; Fukuta, Y.; Sasaki, T.; Yano, M. An SNP caused loss of seed shattering during rice domestication. Science 2006, 312, 1392–1406. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.W.; Griffith, M.E.; Li, X.R.; Zhu, Z.F.; Tan, L.B.; Fu, Y.C.; Zhang, W.X.; Wang, X.K.; Xie, D.X.; Sun, C.Q. Origin of seed shattering in rice (Oryza sativa L.). Planta 2007, 226, 11–20. [Google Scholar] [CrossRef]
- Ross-Ibarra, J.; Morrell, P.; Gaut, B. Plant domestication: A unique opportunity to identify the genetic basis of adaptation. Proc. Natl. Acad. Sci. USA 2007, 104, 8641–8648. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.B.; Zhu, Q.H.; Wu, Z.Q.; Ross-Ibarra, J.; Gaut, B.; Song, G.; Sang, T. Selection on grain shattering genes and rates of rice domestication. New Phytol. 2009, 184, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [PubMed] [Green Version]
- Feng, Q.; Zhang, Y.J.; Hao, P.; Wang, S.Y.; Fu, G.; Huang, Y.C.; Li, Y.; Zhu, J.J.; Liu, Y.L.; Hu, X.; et al. Sequence and analysis of rice chromosome 4. Nature 2002, 420, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wu, J.; Kanamori, H.; Katayose, Y.; Fujisawa, M.; Namiki, N.; Mizuno, H.; Yamamoto, K.; Antonio, B.; Baba, T.; et al. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar]
- Thompson, J.; Gibson, T.; Plewniak, F.; Jeanmougin, F.; Higgins, D. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1998, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- McCouch, S.; Teytelman, L.; Xu, Y.B.; Lobos, K.; Clare, K.; Walton, M.; Fu, B.; Maghirang, R.; Li, Z.K.; Xing, Y.Z.; et al. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res. 2003, 9, 199–207. [Google Scholar] [CrossRef]
- Lu, B.-R.; Cai, X.X.; Jin, X. Efficient indica and japonica rice identification based on the InDel molecular method: Its implication in rice breeding and evolutionary research. Prog. Nat. Sci. 2009, 19, 1241–1252. [Google Scholar] [CrossRef]
- Liu, P.; Cai, X.X.; Lu, B.-R. Single-seeded InDel fingerprints in rice: An effective tool for indica-japonica rice classification and evolutionary studies. J. Syst. Evol. 2012, 50, 1–11. [Google Scholar] [CrossRef]
- Librado, P.J.R.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1502. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.J.; Donnelly, P.J. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.S.; Regnaut, S.J.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Reagon, M.; Thurber, C.S.; Gross, B.L.; Olsen, K.M.; Jia, Y.; Caicedo, A.L. Genomic patterns of nucleotide diversity in divergent populations of U.S. weedy rice. BMC Evol. Biol. 2010, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Zhou, Y.J.; Mao, L.F.; Ye, C.; Wang, W.D.; Zhang, J.P.; Yu, Y.Y.; Fu, F.; Wang, Y.F.; Qian, F.J.; et al. Genomic variation associated with local adaptation of weedy rice during de-domestication. Nat. Commun. 2017, 8, e15323. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Qian, Q.; Ma, D.; Xu, Z.; Liu, D.; Du, H.; Chen, W. Introgression and selection shaping the genome and adaptive loci of weedy rice in northern China. New Phytol. 2013, 197, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Mccouch, S. The complex history of the domestication of rice. Ann. Bot. 2007, 100, 951–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reig Valiente, J.; Viruel, J.; Sales, E.; Marqués, L.; Terol, J.; Gut, M.; Derdak, S.; Talon, M.; Domingo, C. Genetic diversity and population structure of rice varieties cultivated in temperate regions. Rice 2016, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.S.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.S.; Wu, Z.C.; Li, M.; Zheng, T.Q.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Gao, L.Z.; Zhang, C.H.; Chang, L.P.; Jia, J.Z.; Qiu, Z.E.; Dong, Y.S. Microsatellite diversity within Oryza sativa with emphasis on indica–japonica divergence. Genet. Res. 2005, 85, 1–14. [Google Scholar] [CrossRef]
- Garris, A.; Tai, T.; Coburn, J.; Kresovich, S.; Mccouch, S. Genetic structure and diversity in Oryza sativa L. Genetics 2005, 169, 1631–1638. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.J.; Wang, Z.; Feng, Y.; Yao, N.; Yang, J.; Lu, B.-R. Genetic divergence of weedy rice populations associated with their geographic location and coexisting conspecific crop: Implications on adaptive evolution of agricultural weeds. J. Syst. Evol. 2015, 53, 330–338. [Google Scholar] [CrossRef]
- Song, D.Y.; Wang, Z.; Song, Z.J.; Zhou, C.C.; Xu, P.H.; Yang, J.; Yang, J.; Lu, B.-R. Increased novel single nucleotide polymorphisms in weedy rice populations associated with the change of farming styles: Implications in adaptive mutation and evolution. J. Syst. Evol. 2017, 55, 149–157. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, L.; Wang, Z.; Lu, B.-R. Non-random transmission of parental alleles into crop-wild and crop-weed hybrid lineages separated by a transgene and neutral identifiers in rice. Sci. Rep. 2017, 7, e10436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellstrand, N.C. Is gene flow the most important evolutionary force in plants? Am. J. Bot. 2014, 101, 737–753. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Taxon | No. of Variable Sites | No. of Haplotypes | Proportion of Total Haplotypes 1 | Type of Haplotypes | |
|---|---|---|---|---|---|---|
| Shared Haplotypes | Specific Haplotypes 2 | |||||
| sh4 | Wild rice | 101 | 103 | 87.3% | Hs1, Hs2 | Hs3~Hs103 |
| Weedy rice | 35 | 9 | 7.6% | Hs1, Hs2 | Hs104~Hs110 | |
| Cultivated rice | 4 | 6 | 5.1% | Hs1, Hs2 | Hs111~Hs114 | |
| qsh1 | Wild rice | 70 | 54 | 57.4% | Hq1~Hq4 | Hq5~Hq54 |
| Weedy rice | 33 | 25 | 26.6% | Hq1~Hq4 | Hq5, Hq8, Hq9, Hq55~Hq72 | |
| Cultivated rice | 24 | 15 | 16.0% | Hq1~Hq4 | Hq55~Hq57, Hq73~Hq80 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.-Q.; Fang, J.; Wang, Y.; Pang, L.-H.; Lu, B.-R. Key Roles of De-Domestication and Novel Mutation in Origin and Diversification of Global Weedy Rice. Biology 2021, 10, 828. https://doi.org/10.3390/biology10090828
Zhu Y-Q, Fang J, Wang Y, Pang L-H, Lu B-R. Key Roles of De-Domestication and Novel Mutation in Origin and Diversification of Global Weedy Rice. Biology. 2021; 10(9):828. https://doi.org/10.3390/biology10090828
Chicago/Turabian StyleZhu, Yong-Qing, Jia Fang, Ying Wang, Li-Hao Pang, and Bao-Rong Lu. 2021. "Key Roles of De-Domestication and Novel Mutation in Origin and Diversification of Global Weedy Rice" Biology 10, no. 9: 828. https://doi.org/10.3390/biology10090828
APA StyleZhu, Y.-Q., Fang, J., Wang, Y., Pang, L.-H., & Lu, B.-R. (2021). Key Roles of De-Domestication and Novel Mutation in Origin and Diversification of Global Weedy Rice. Biology, 10(9), 828. https://doi.org/10.3390/biology10090828