
Genetic Mutations and Non-Coding RNA-Based Epigenetic Alterations Mediating the Warburg Effect in Colorectal Carcinogenesis

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
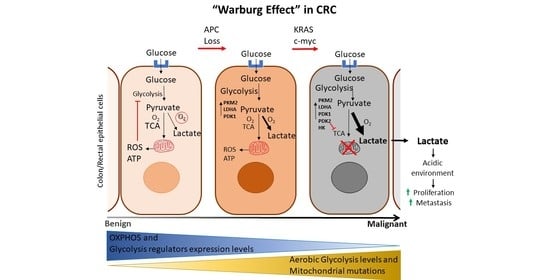
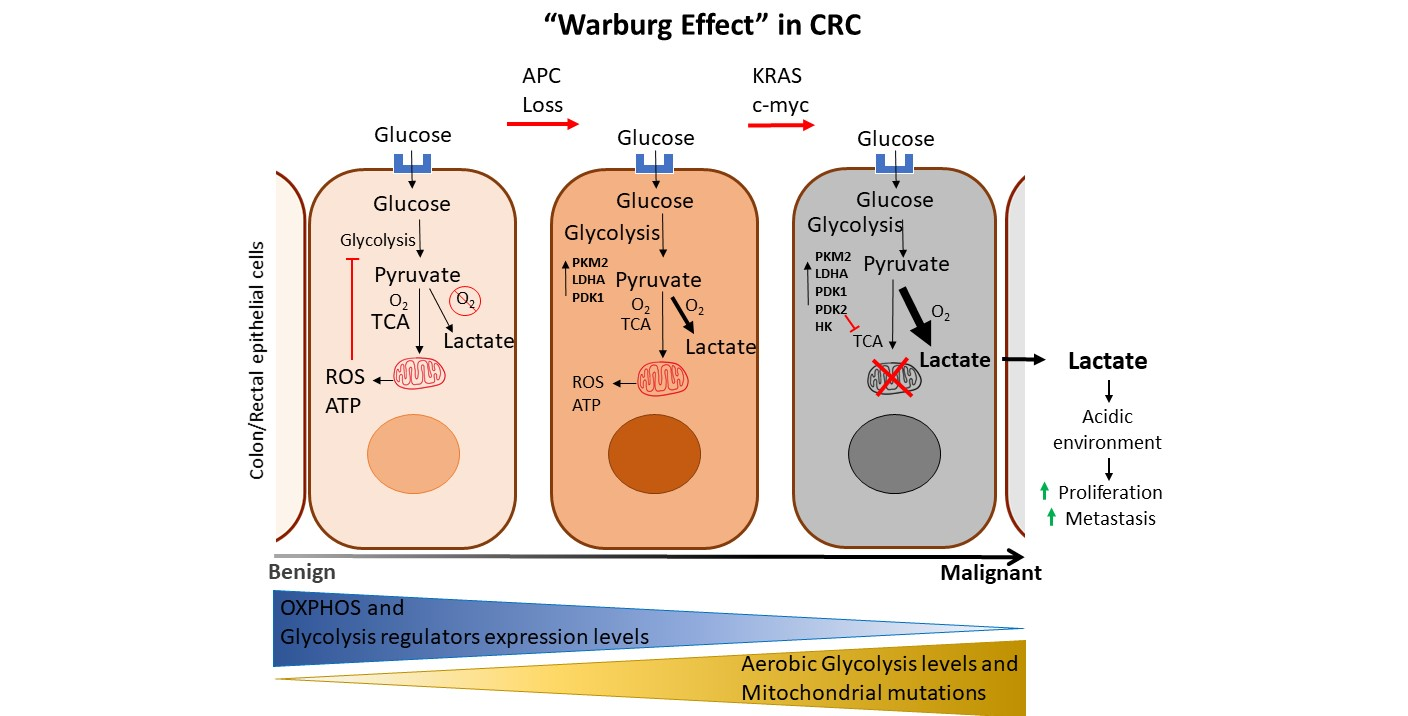
1. Introduction
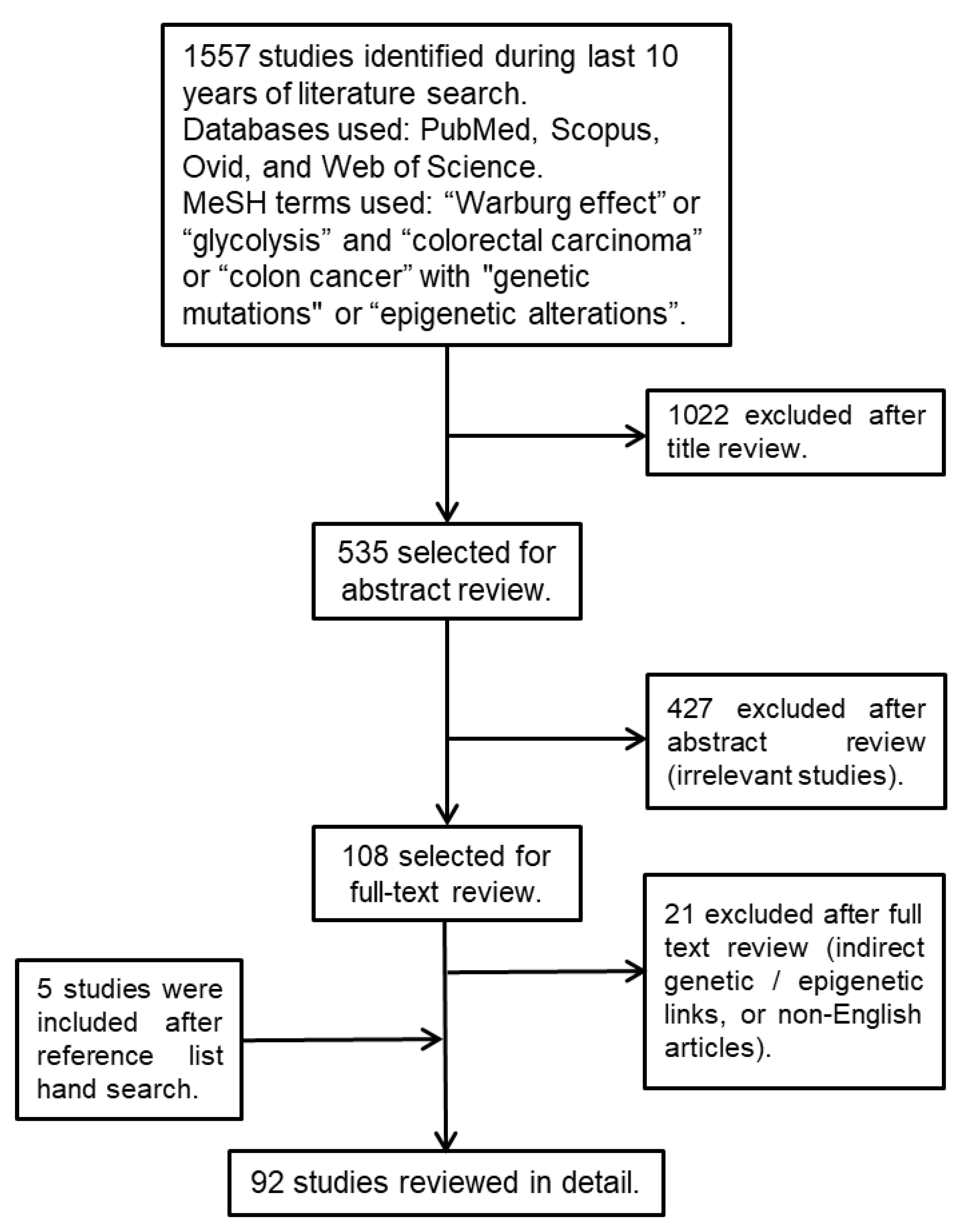
2. Materials and Methods
3. Metabolic Reprogramming in CRC: Genetic Mutations and ncRNA-Mediated Epigenetic Alteration
3.1. Oncogenes and Tumor Suppressor Genes
3.1.1. KRAS Proto-Oncogene
3.1.2. c-Myc Proto-Oncogene
3.1.3. Hypoxia-Inducible Factor 1-Alpha (HIF1-α)
3.1.4. Adenomatous Polyposis Coli (APC)
3.1.5. TP53
3.2. Key Enzymes and Transporters Involved in the Warburg Effect
3.2.1. Mitochondrial Pyruvate Carrier (MPC)
3.2.2. Pyruvate Kinase Isozyme M2 (PKM2)
3.2.3. Hexokinase 2 (HK2)
3.2.4. Glucose Transporter 1 (GLUT1)
4. Conclusions and Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Mao, T.; Li, S.; He, J.; Hou, X.; Li, H.; Zhan, M.; Yang, X.; Li, R.; Xiao, J.; et al. DT-13 inhibited the proliferation of colorectal cancer via glycolytic metabolism and AMPK/mTOR signaling pathway. Phytomedicine 2019, 54, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, J.; Xu, Q.; Duan, W.; Yang, L.; Wu, X.; Lu, G.; Zhang, L.; Zheng, Y. Oxymatrine Inhibits Colorectal Cancer Metastasis via Attenuating PKM2-Mediated Aerobic Glycolysis. Cancer Manag. Res. 2020, 12, 9503–9513. [Google Scholar] [CrossRef] [PubMed]
- Eslami, M.; Sadrifar, S.; Karbalaei, M.; Keikha, M.; Kobyliak, N.M.; Yousefi, B. Importance of the Microbiota Inhibitory Mechanism on the Warburg Effect in Colorectal Cancer Cells. J. Gastrointest. Cancer 2020, 51, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Dai, W.; Meng, X.; Mo, S.; Xiang, W.; Xu, Y.; Zhang, L.; Wang, R.; Li, Q.; Cai, G. FOXE1 represses cell proliferation and Warburg effect by inhibiting HK2 in colorectal cancer. Cell Commun. Signal. 2020, 18, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Chekulayev, V.; Mado, K.; Shevchuk, I.; Koit, A.; Kaldma, A.; Klepinin, A.; Timohhina, N.; Tepp, K.; Kandashvili, M.; Ounpuu, L.; et al. Metabolic remodeling in human colorectal cancer and surrounding tissues: Alterations in regulation of mitochondrial respiration and metabolic fluxes. Biochem. Biophys. Reports 2015, 4, 111–125. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Kaldma, A.; Klepinin, A.; Chekulayev, V.; Mado, K.; Shevchuk, I.; Timohhina, N.; Tepp, K.; Kandashvili, M.; Varikmaa, M.; Koit, A.; et al. An in situ study of bioenergetic properties of human colorectal cancer: The regulation of mitochondrial respiration and distribution of flux control among the components of ATP synthasome. Int. J. Biochem. Cell Biol. 2014, 55, 171–186. [Google Scholar] [CrossRef]
- Smith, A.L.; Whitehall, J.C.; Bradshaw, C.; Gay, D.; Robertson, F.; Blain, A.P.; Hudson, G.; Pyle, A.; Houghton, D.; Hunt, M.; et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 2020, 1, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Shen, Z.-Y.; Zhan, Y.-Z.; Feng, X.-C.; Chen, K.-L.; Li, Y.-S.; Deng, H.-J.; Pan, S.-M.; Wu, D.-H.; Ding, Y. CD36 inhibits β-catenin/c-myc-mediated glycolysis through ubiquitination of GPC4 to repress colorectal tumorigenesis. Nat. Commun. 2019, 10, 3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebane-Klemm, E.; Truu, L.; Reinsalu, L.; Puurand, M.; Shevchuk, I.; Chekulayev, V.; Timohhina, N.; Tepp, K.; Bogovskaja, J.; Afanasjev, V.; et al. Mitochondrial Respiration in KRAS and BRAF Mutated Colorectal Tumors and Polyps. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, A.; Boland, C.R. Epigenetics of colorectal cancer. Gastroenterology 2012, 143, 1442–1460.e1. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, J.S.; Eladl, M.A.; Khoder, G. Helicobacter pylori-induced DNA Methylation as an Epigenetic Modulator of Gastric Cancer: Recent Outcomes and Future Direction. Pathogens 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ma, R.; Wu, Y.; Zhai, Y.; Li, S. Reciprocal Regulation of Metabolic Reprogramming and Epigenetic Modifications in Cancer. Front. Genet. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- El Halabi, I.; Bejjany, R.; Nasr, R.; Mukherji, D.; Temraz, S.; Nassar, F.J.; El Darsa, H.; Shamseddine, A. Ascorbic Acid in Colon Cancer: From the Basic to the Clinical Applications. Int. J. Mol. Sci. 2018, 19, 2752. [Google Scholar] [CrossRef] [Green Version]
- Kawada, K.; Toda, K.; Sakai, Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int. J. Clin. Oncol. 2017, 22, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Huang, W.; Ge, X.; Xue, L.; Zhao, W.; Xue, J. Type Iγ phosphatidylinositol phosphate kinase promotes tumor growth by facilitating Warburg effect in colorectal cancer. EBioMedicine 2019, 44, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, T.; Sun, H.; Weng, W.; Zhang, Q.; Liu, C.; Han, Y.; Sheng, W. Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. Cancer Sci. 2018, 109, 1468–1479. [Google Scholar] [CrossRef] [Green Version]
- Cha, P.-H.; Hwang, J.-H.; Kwak, D.-K.; Koh, E.; Kim, K.-S.; Choi, K.-Y. APC loss induces Warburg effect via increased PKM2 transcription in colorectal cancer. Br. J. Cancer 2021, 124, 634–644. [Google Scholar] [CrossRef]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2020, 39, 469–485. [Google Scholar] [CrossRef]
- Papageorgis, P.; Cheng, K.; Ozturk, S.; Gong, Y.; Lambert, A.W.; Abdolmaleky, H.M.; Zhou, J.-R.; Thiagalingam, S. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011, 71, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Fang, J.; Wang, S.; Deng, B.; Zhu, L. MicroRNA-135b regulates the stability of PTEN and promotes glycolysis by targeting USP13 in human colorectal cancers. Oncol. Rep. 2015, 33, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yan, T.; Bao, Y.; Shen, C.; Yu, C.; Zhu, X.; Tian, X.; Guo, F.; Liang, Q.; Liu, Q.; et al. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nat. Commun. 2019, 10, 3499. [Google Scholar] [CrossRef]
- Liu, F.; Di Wang, X. miR-150-5p represses TP53 tumor suppressor gene to promote proliferation of colon adenocarcinoma. Sci. Rep. 2019, 9, 6740. [Google Scholar] [CrossRef]
- Fang, S.; Fang, X. Advances in glucose metabolism research in colorectal cancer. Biomed. Rep. 2016, 5, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.E.; Short, S.P.; Williams, C.S. Colorectal Cancer and Metabolism. Curr. Colorectal Cancer Rep. 2018, 14, 226–241. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, Q.; Yuan, W.; Li, X.; Chen, C.; Guo, Y.; Shao, B.; Dang, Q.; Zhou, Q.; Wang, Q.; et al. MiR-103a-3p promotes tumour glycolysis in colorectal cancer via hippo/YAP1/HIF1A axis. J. Exp. Clin. Cancer Res. 2020, 39, 250. [Google Scholar] [CrossRef]
- Nie, H.; Ju, H.; Fan, J.; Shi, X.; Cheng, Y.; Cang, X.; Zheng, Z.; Duan, X.; Yi, W. O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth. Nat. Commun. 2020, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xiang, F.; Huang, Y.; Shi, L.; Hu, C.; Yang, Y.; Wang, D.; He, N.; Tao, K.; Wu, K.; et al. Interleukin-22 promotes aerobic glycolysis associated with tumor progression via targeting hexokinase-2 in human colon cancer cells. Oncotarget 2017, 8, 25372–25383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, J.S.; Guimei, M.; Jayakumar, M.N.; Shafarin, J.; Janeeh, A.S.; AbuJabal, R.; Eladl, M.A.; Ranade, A.V.; Ali, A.; Hamad, M. Estrogen-induced hypomethylation and overexpression of YAP1 facilitate breast cancer cell growth and survival. Neoplasia 2021, 23, 68–79. [Google Scholar] [CrossRef]
- Muhammad, J.S.; Bajbouj, K.; Shafarin, J.; Hamad, M. Estrogen-induced epigenetic silencing of FTH1 and TFRC genes reduces liver cancer cell growth and survival. Epigenetics 2020, 15, 1302–1318. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Faraonio, R.; Buonaguro, L.; Annunziata, C.; Starita, N.; Cerasuolo, A.; Pezzuto, F.; Tornesello, A.L.; Buonaguro, F.M. The Role of microRNAs, Long Non-coding RNAs, and Circular RNAs in Cervical Cancer. Front. Oncol. 2020, 10, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusic, M.; Prokisch, H. ncRNAs: New Players in Mitochondrial Health and Disease? Front. Genet. 2020, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; Yu, W. Long noncoding RNA KCNQ1OT1 promotes colorectal carcinogenesis by enhancing aerobic glycolysis via hexokinase-2. Aging 2020, 12, 11685–11697. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Chen, J.; Zhang, X.; Wang, Z.; Chen, J.; Lin, X.; Huang, H.; Fu, W.; Liang, J.; Wu, W.; et al. The HIF-1α antisense long non-coding RNA drives a positive feedback loop of HIF-1α mediated transactivation and glycolysis. Nat. Commun. 2021, 12, 1341. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Bao, J.; Feng, J.-F. Long non-coding RNA BCYRN1 exerts an oncogenic role in colorectal cancer by regulating the miR-204-3p/KRAS axis. Cancer Cell Int. 2020, 20, 453. [Google Scholar] [CrossRef]
- Pancione, M.; Remo, A.; Colantuoni, V. Genetic and Epigenetic Events Generate Multiple Pathways in Colorectal Cancer Progression. Patholog. Res. Int. 2012, 2012, 509348. [Google Scholar] [CrossRef] [Green Version]
- Dela Cruz, M.; Ledbetter, S.; Chowdhury, S.; Tiwari, A.K.; Momi, N.; Wali, R.K.; Bliss, C.; Huang, C.; Lichtenstein, D.; Bhattacharya, S.; et al. Metabolic reprogramming of the premalignant colonic mucosa is an early event in carcinogenesis. Oncotarget 2017, 8, 20543–20557. [Google Scholar] [CrossRef] [PubMed]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef] [Green Version]
- Dinu, D.; Dobre, M.; Panaitescu, E.; Bîrlă, R.; Iosif, C.; Hoara, P.; Caragui, A.; Boeriu, M.; Constantinoiu, S.; Ardeleanu, C. Prognostic significance of KRAS gene mutations in colorectal cancer--preliminary study. J. Med. Life 2014, 7, 581–587. [Google Scholar] [PubMed]
- Aguilera, O.; Serna-Blasco, R. Targeting KRAS Mutant CMS3 Subtype by Metabolic Inhibitors. Adv. Exp. Med. Biol. 2018, 1110, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, B.; Biechl, P.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; Schwaiger, M.; Eisenreich, W. Diverse metabolic response of cancer cells treated with a 213Bi-anti-EGFR-immunoconjugate. Sci. Rep. 2021, 11, 6227. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.-H.; Lee, E.J.; Park, J.W.; Lee, J.H.; Moon, S.H.; Cho, Y.S.; Lee, K.-H. EGF receptor stimulation shifts breast cancer cell glucose metabolism toward glycolytic flux through PI3 kinase signaling. PLoS ONE 2019, 14, e0221294. [Google Scholar] [CrossRef]
- Ye, H.; Liu, Y.; Wu, K.; Luo, H.; Cui, L. AMPK activation overcomes anti-EGFR antibody resistance induced by KRAS mutation in colorectal cancer. Cell Commun. Signal. 2020, 18, 115. [Google Scholar] [CrossRef]
- Bellier, J.; Nokin, M.-J.; Caprasse, M.; Tiamiou, A.; Blomme, A.; Scheijen, J.L.; Koopmansch, B.; MacKay, G.M.; Chiavarina, B.; Costanza, B.; et al. Methylglyoxal Scavengers Resensitize KRAS-Mutated Colorectal Tumors to Cetuximab. Cell Rep. 2020, 30, 1400–1416. [Google Scholar] [CrossRef]
- Zhu, L.-L.; Wu, Z.; Li, R.-K.; Xing, X.; Jiang, Y.-S.; Li, J.; Wang, Y.-H.; Hu, L.-P.; Wang, X.; Qin, W.-T.; et al. Deciphering the genomic and lncRNA landscapes of aerobic glycolysis identifies potential therapeutic targets in pancreatic cancer. Int. J. Biol. Sci. 2021, 17, 107–118. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Lan, Z. Silencing UNC5B antisense lncRNA 1 represses growth and metastasis of human Colon cancer cells via raising miR-622. Artif. Cells Nanomed. Biotechnol. 2020, 48, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- He, W.-L.; Weng, X.-T.; Wang, J.-L.; Lin, Y.-K.; Liu, T.-W.; Zhou, Q.-Y.; Hu, Y.; Pan, Y.; Chen, X.-L. Association between c-Myc and Colorectal Cancer Prognosis: A Meta-Analysis. Front. Physiol. 2018, 9, 1549. [Google Scholar] [CrossRef] [Green Version]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Matsuhashi, N.; Futamura, M.; Ito, Y.; Otsuki, Y.; Yoshida, K.; et al. Positive feedback of DDX6/c-Myc/PTB1 regulated by miR-124 contributes to maintenance of the Warburg effect in colon cancer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sun, Y.; Fei, Z.; Yang, Z.; Duan, K.; Zi, J.; Cui, Q.; Yu, M.; Xiong, W. Leptin promotes fatty acid oxidation and OXPHOS via the c-Myc/PGC-1 pathway in cancer cells. Acta Biochim. Biophys. Sin. 2019, 51, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Wu, L.; Wang, Y.; Yuan, X. Long Non-coding RNA MEG3 Activated by Vitamin D Suppresses Glycolysis in Colorectal Cancer via Promoting c-Myc Degradation. Front. Oncol. 2020, 10, 274. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhu, Y.; Hong, X.; Zhang, M.; Qiu, X.; Wang, Z.; Qi, Z.; Hong, X. miR-181d and c-myc-mediated inhibition of CRY2 and FBXL3 reprograms metabolism in colorectal cancer. Cell Death Dis. 2017, 8, e2958. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Duan, W.; Liu, Y. Ketamine inhibits aerobic glycolysis in colorectal cancer cells by blocking the NMDA receptor-CaMK II-c-Myc pathway. Clin. Exp. Pharmacol. Physiol. 2020, 47, 848–856. [Google Scholar] [CrossRef]
- Wu, Z.; Han, X.; Tan, G.; Zhu, Q.; Chen, H.; Xia, Y.; Gong, J.; Wang, Z.; Wang, Y.; Yan, J. Dioscin Inhibited Glycolysis and Induced Cell Apoptosis in Colorectal Cancer via Promoting c-myc Ubiquitination and Subsequent Hexokinase-2 Suppression. Oncol. Targets Ther. 2020, 13, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Sun, S.; Xia, C.; Xu, Y. HIF-1α induced lncRNA LINC00511 accelerates the colorectal cancer proliferation through positive feedback loop. Biomed. Pharmacother. 2020, 125, 110014. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, H.; Wang, F.; Han, Y.; Yang, W. Pim-2 Modulates Aerobic Glycolysis and Energy Production during the Development of Colorectal Tumors. Int. J. Med. Sci. 2015, 12, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, E.; Gouw, A.S.H.; Posthumus, M.D.; van Leeuwen, M.A.; Boerboom, A.L.; Bijzet, J.; Bos, R.; Limburg, P.C.; Kallenberg, C.G.M.; Westra, J. Hypoxia inducible factor-1-alpha (HIF-1alpha) is related to both angiogenesis and inflammation in rheumatoid arthritis. Clin. Exp. Rheumatol. 2009, 27, 945–951. [Google Scholar] [PubMed]
- Xu, Y.; Han, S.; Lei, K.; Chang, X.; Wang, K.; Li, Z.; Liu, J. Anti-Warburg effect of rosmarinic acid via miR-155 in colorectal carcinoma cells. Eur. J. Cancer Prev. 2016, 25, 481–489. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Kuranaga, Y.; Tahara, T.; Yamashita, H.; Shibata, T.; Nagasaka, M.; Funasaka, K.; Ohmiya, N.; Akao, Y. Induced miR-31 by 5-fluorouracil exposure contributes to the resistance in colorectal tumors. Cancer Sci. 2019, 110, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Quan, Y.; Chen, Y.; Huang, Y.; Huang, R.; Yu, W.; Wu, D.; Ye, M.; Min, Z.; Yu, B. Knockdown of RNA N6-methyladenosine methyltransferase METTL3 represses Warburg effect in colorectal cancer via regulating HIF-1α. Signal Transduct. Targets Ther. 2021, 6, 89. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Zhuo, M.; Li, M.; Chen, Q.; Mo, P.; Yu, C. Histone demethylase JMJD2D activates HIF1 signaling pathway via multiple mechanisms to promote colorectal cancer glycolysis and progression. Oncogene 2020, 39, 7076–7091. [Google Scholar] [CrossRef]
- Cruz-Gil, S.; Sánchez-Martínez, R.; Wagner-Reguero, S.; Stange, D.; Schölch, S.; Pape, K.; Ramírez de Molina, A. A more physiological approach to lipid metabolism alterations in cancer: CRC-like organoids assessment. PLoS ONE 2019, 14, e0219944. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sviripa, V.M.; Xie, Y.; Yu, T.; Haney, M.G.; Blackburn, J.S.; Adeniran, C.A.; Zhan, C.-G.; Watt, D.S.; Liu, C. Epigenetic Regulation of Wnt Signaling by Carboxamide-Substituted Benzhydryl Amines that Function as Histone Demethylase Inhibitors. iScience 2020, 23, 101795. [Google Scholar] [CrossRef]
- Pan, J.-H.; Zhou, H.; Zhu, S.-B.; Huang, J.-L.; Zhao, X.-X.; Ding, H.; Qin, L.; Pan, Y.-L. Nicotinamide phosphoribosyl transferase regulates cell growth via the Sirt1/P53 signaling pathway and is a prognosis marker in colorectal cancer. J. Cell. Physiol. 2019, 234, 4385–4395. [Google Scholar] [CrossRef]
- Ohashi, T.; Eguchi, H.; Kawamoto, K.; Konno, M.; Asai, A.; Colvin, H.; Ueda, Y.; Takaoka, H.; Iwagami, Y.; Yamada, D.; et al. Mitochondrial pyruvate carrier modulates the epithelial-mesenchymal transition in cholangiocarcinoma. Oncol. Rep. 2018, 39, 1276–1282. [Google Scholar] [CrossRef] [Green Version]
- Bensard, C.L.; Wisidagama, D.R.; Olson, K.A.; Berg, J.A.; Krah, N.M.; Schell, J.C.; Nowinski, S.M.; Fogarty, S.; Bott, A.J.; Wei, P.; et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab. 2020, 31, 284.e7–300.e7. [Google Scholar] [CrossRef]
- Schell, J.C.; Olson, K.A.; Jiang, L.; Hawkins, A.J.; Van Vranken, J.G.; Xie, J.; Egnatchik, R.A.; Earl, E.G.; DeBerardinis, R.J.; Rutter, J. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 2014, 56, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.; Li, Q.; Huang, L.; Li, D.; Li, X. Sirt3 binds to and deacetylates mitochondrial pyruvate carrier 1 to enhance its activity. Biochem. Biophys. Res. Commun. 2015, 468, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, Y.; Konno, M.; Koseki, J.; Colvin, H.; Asai, A.; Tamari, K.; Satoh, T.; Mori, M.; Doki, Y.; Ogawa, K.; et al. Mitochondrial pyruvate carrier 1 expression controls cancer epithelial-mesenchymal transition and radioresistance. Cancer Sci. 2019, 110, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Zhao, X.; Huang, L.; Zhang, T.; Yang, F.; Xie, L.; Song, S.; Miao, P.; Zhao, L.; Sun, X.; et al. Proviral insertion in murine lymphomas 2 (PIM2) oncogene phosphorylates pyruvate kinase M2 (PKM2) and promotes glycolysis in cancer cells. J. Biol. Chem. 2013, 288, 35406–35416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Sakai, M.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakayama, T.; Ueda, H.; Nakagawa, Y.; Ito, Y.; et al. PTBP1-associated microRNA-1 and -133b suppress the Warburg effect in colorectal tumors. Oncotarget 2016, 7, 18940–18952. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhao, X.; Zhou, Y.; Hu, Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 2012, 28, 1346–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Yang, P.; Amin, S.; Li, Z. A novel miR-206/hnRNPA1/PKM2 axis reshapes the Warburg effect to suppress colon cancer growth. Biochem. Biophys. Res. Commun. 2020, 531, 465–471. [Google Scholar] [CrossRef]
- Bian, Z.; Zhang, J.; Li, M.; Feng, Y.; Wang, X.; Zhang, J.; Yao, S.; Jin, G.; Du, J.; Han, W.; et al. LncRNA-FEZF1-AS1 Promotes Tumor Proliferation and Metastasis in Colorectal Cancer by Regulating PKM2 Signaling. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4808–4819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, H.; Yang, H.; Bai, M.; Ning, T.; Deng, T.; Liu, R.; Fan, Q.; Zhu, K.; Li, J.; et al. Exosome-delivered circRNA promotes glycolysis to induce chemoresistance through the miR-122-PKM2 axis in colorectal cancer. Mol. Oncol. 2020, 14, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cao, L.; Tian, Y.; Zhang, P.; Ding, C.; Lu, W.; Jia, C.; Shao, C.; Liu, W.; Wang, D.; et al. Butyrate Suppresses the Proliferation of Colorectal Cancer Cells via Targeting Pyruvate Kinase M2 and Metabolic Reprogramming. Mol. Cell. Proteom. 2018, 17, 1531–1545. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Ma, Y.; Cao, L.; Zhan, S.; Xu, Y.; Fu, F.; Liu, C.; Zhang, G.; Wang, Z.; Wang, R.; et al. B7-H3 promotes aerobic glycolysis and chemoresistance in colorectal cancer cells by regulating HK2. Cell Death Dis. 2019, 10, 308. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.; Coomber, B.L. Hexokinase II expression is correlated with colorectal cancer prognosis. Cancer Treat. Commun. 2016, 6, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Kudryavtseva, A.V.; Fedorova, M.S.; Zhavoronkov, A.; Moskalev, A.A.; Zasedatelev, A.S.; Dmitriev, A.A.; Sadritdinova, A.F.; Karpova, I.Y.; Nyushko, K.M.; Kalinin, D.V.; et al. Effect of lentivirus-mediated shRNA inactivation of HK1, HK2, and HK3 genes in colorectal cancer and melanoma cells. BMC Genet. 2016, 17, 156. [Google Scholar] [CrossRef] [Green Version]
- Gregersen, L.H.; Jacobsen, A.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer 2012, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Kawada, K.; Iwamoto, M.; Sakai, Y. Mechanisms underlying (18)F-fluorodeoxyglucose accumulation in colorectal cancer. World J. Radiol. 2016, 8, 880–886. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Zhang, Y.-H.; Qin, X.-J.; Wang, Y.-X.; Fu, J. Circular RNA circDENND4C facilitates proliferation, migration and glycolysis of colorectal cancer cells through miR-760/GLUT1 axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2387–2400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, Y.; Liu, F.; Yin, M. Overexpression of miRNA-143 Inhibits Colon Cancer Cell Proliferation by Inhibiting Glucose Uptake. Arch. Med. Res. 2018, 49, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gao, S.; Liu, W.; Wong, C.-C.; Wu, J.; Wu, J.; Liu, D.; Gou, H.; Kang, W.; Zhai, J.; et al. RNA N(6)-Methyladenosine Methyltransferase METTL3 Facilitates Colorectal Cancer by Activating the m(6)A-GLUT1-mTORC1 Axis and Is a Therapeutic Target. Gastroenterology 2021, 160, 1284–1300. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Xie, F.; Li, M.; Liang, Z.; Xu, W.; Yang, J.; Liu, C.; Li, H.; Zhou, H.; Qu, L.-H. Oridonin induces autophagy via inhibition of glucose metabolism in p53-mutated colorectal cancer cells. Cell Death Dis. 2017, 8, e2633. [Google Scholar] [CrossRef]
- Geng, H.-W.; Yin, F.-Y.; Zhang, Z.-F.; Gong, X.; Yang, Y. Butyrate Suppresses Glucose Metabolism of Colorectal Cancer Cells via GPR109a-AKT Signaling Pathway and Enhances Chemotherapy. Front. Mol. Biosci. 2021, 8, 634874. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Genes | Function | Epigenetic Alteration | Therapy Resistance | Anti-Glycolysis Therapy | ||
|---|---|---|---|---|---|---|
| Molecule | Expression in CRC | Effect on Glycolysis | ||||
| KRAS | Oncogenic activator of RAS/MAPK | UNC5B-AS1 [50] | Upregulated | Activating | Anti-EGFR [47,48] | 3-BrPA [19], ascorbic acid [18] |
| c-Myc | Oncogenic Transcription factor | MEG3 [57] GLCC1 [26] miR-181d [58] miR-124 [55] | Downregulated Upregulated Upregulated Downregulated | Inhibitory Activating Activating Inhibitory | N\A | vitamin D [57] Ketamine [59] Dioscin [60] |
| HIF1A | Hypoxia inducible transcription factor | METTL3 [67] YTHDF1 [67] HIFAL [38] | Upregulated Upregulated Upregulated | Activating Activating Inhibitory | 5-FU [66] | Rosmarinic acid [65] |
| APC | Tumor suppressor controlling beta-catenin | N\A | N\A | N\A | DT-13 [2] Metformin [70] | |
| TP53 | Transcription factor and tumor suppresser | N\A | N\A | N\A | DCA [23] FK866 [72] | |
| PKM2 | An enzyme of aerobic glycolysis | miR-1 [82] miR-133b [82] miR-124 [83] miR-137 [83] miR-340 [83] miR-206 [84] MEG3 [57] FEZF1-AS1 [85] miR-122 [86] | Downregulated Downregulated Downregulated Downregulated Downregulated Downregulated Downregulated Upregulated Downregulated | Inhibitory Inhibitory Inhibitory Inhibitory Inhibitory Inhibitory Inhibitory Activating Inhibitory | Oxaliplatin [86] | Butyrate [88] vitamin C [79] Oxymatrine [3] |
| HK2 | An enzyme of aerobic glycolysis | miR-143 [92] MEG3 [57] KCNQ1OT1 [37] | Downregulated Downregulated Upregulated | Inhibitory Inhibitory Activating | Oxaliplatin [89] 5-FU [89] | N\A |
| GLUT1 | Glucose transporter | miR-760 [94] miR-143 [95] circDENND4C [94] METTL3 [96] | Downregulated Downregulated Upregulated Upregulated | Inhibitory Inhibitory Activating Activating | 5-FU [98] | DT-13 [2] Oridonin [97] Oxymatrine [3] Butyrate [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abi Zamer, B.; Abumustafa, W.; Hamad, M.; Maghazachi, A.A.; Muhammad, J.S. Genetic Mutations and Non-Coding RNA-Based Epigenetic Alterations Mediating the Warburg Effect in Colorectal Carcinogenesis. Biology 2021, 10, 847. https://doi.org/10.3390/biology10090847
Abi Zamer B, Abumustafa W, Hamad M, Maghazachi AA, Muhammad JS. Genetic Mutations and Non-Coding RNA-Based Epigenetic Alterations Mediating the Warburg Effect in Colorectal Carcinogenesis. Biology. 2021; 10(9):847. https://doi.org/10.3390/biology10090847
Chicago/Turabian StyleAbi Zamer, Batoul, Wafaa Abumustafa, Mawieh Hamad, Azzam A. Maghazachi, and Jibran Sualeh Muhammad. 2021. "Genetic Mutations and Non-Coding RNA-Based Epigenetic Alterations Mediating the Warburg Effect in Colorectal Carcinogenesis" Biology 10, no. 9: 847. https://doi.org/10.3390/biology10090847
APA StyleAbi Zamer, B., Abumustafa, W., Hamad, M., Maghazachi, A. A., & Muhammad, J. S. (2021). Genetic Mutations and Non-Coding RNA-Based Epigenetic Alterations Mediating the Warburg Effect in Colorectal Carcinogenesis. Biology, 10(9), 847. https://doi.org/10.3390/biology10090847