
New Functions of Vav Family Proteins in Cardiovascular Biology, Skeletal Muscle, and the Nervous System

 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
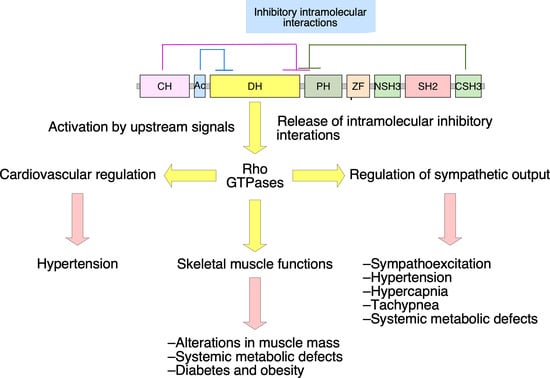
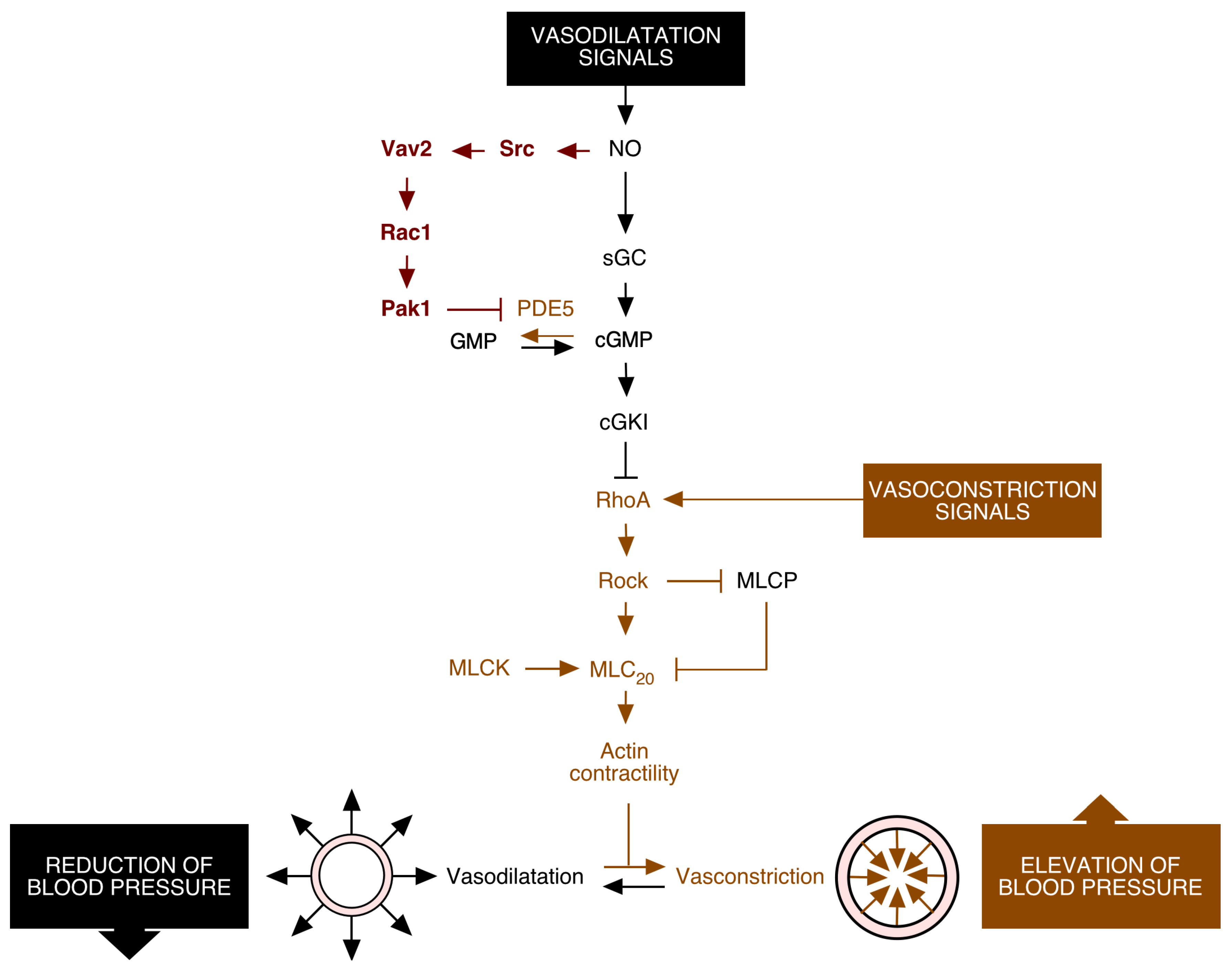
2. Vav2, Vascular Smooth Muscle Cells, and Cardiovascular Regulation
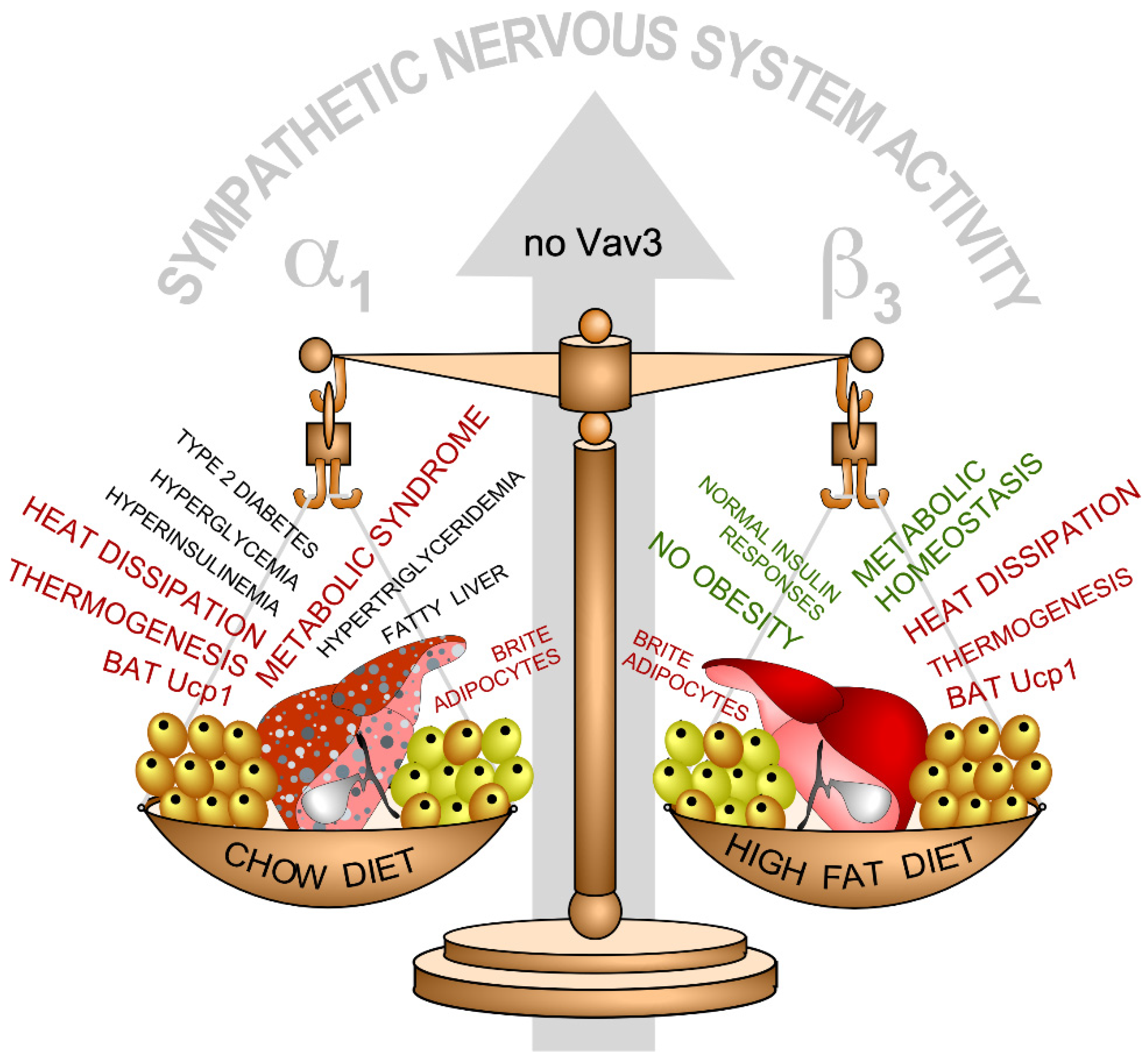
3. Vav2, Skeletal Muscle, and Metabolic Homeostasis
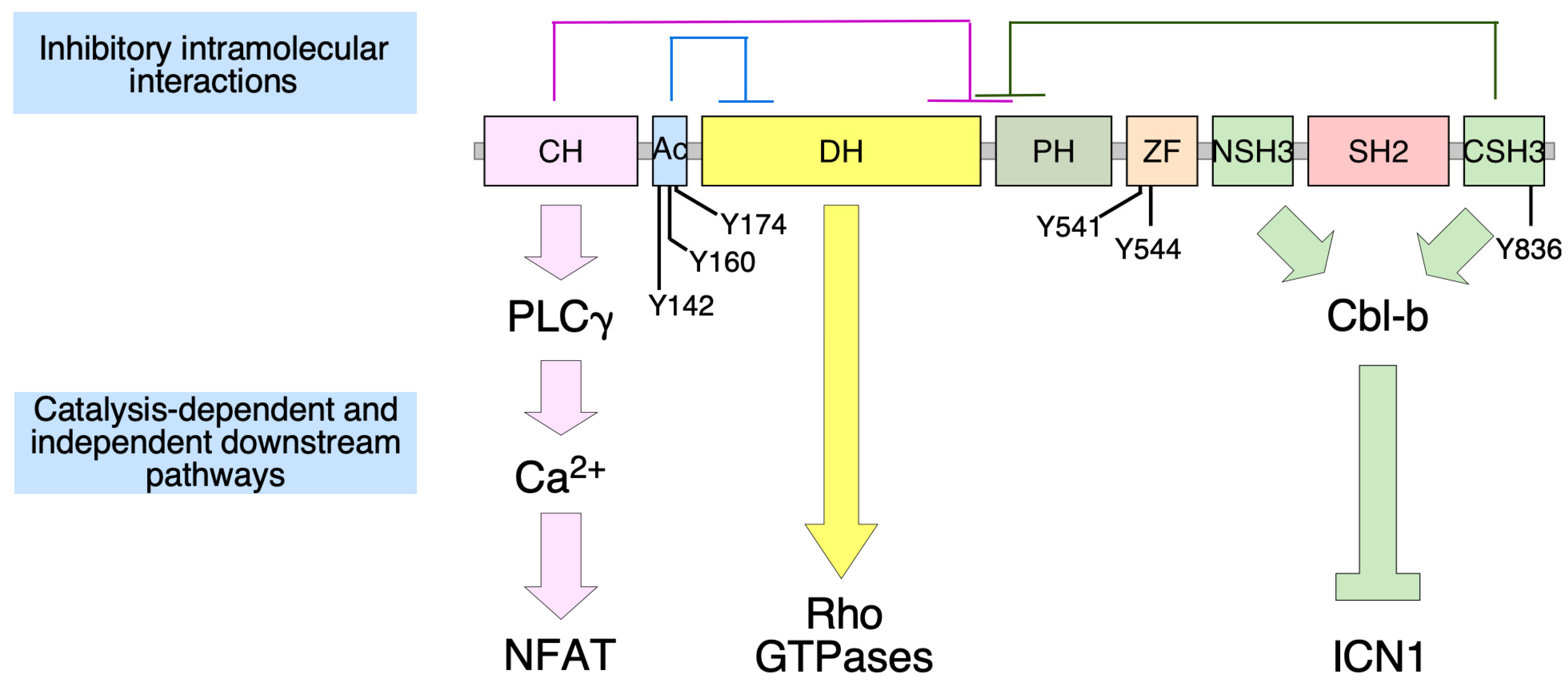
4. Vav2 and Neuronal Functions
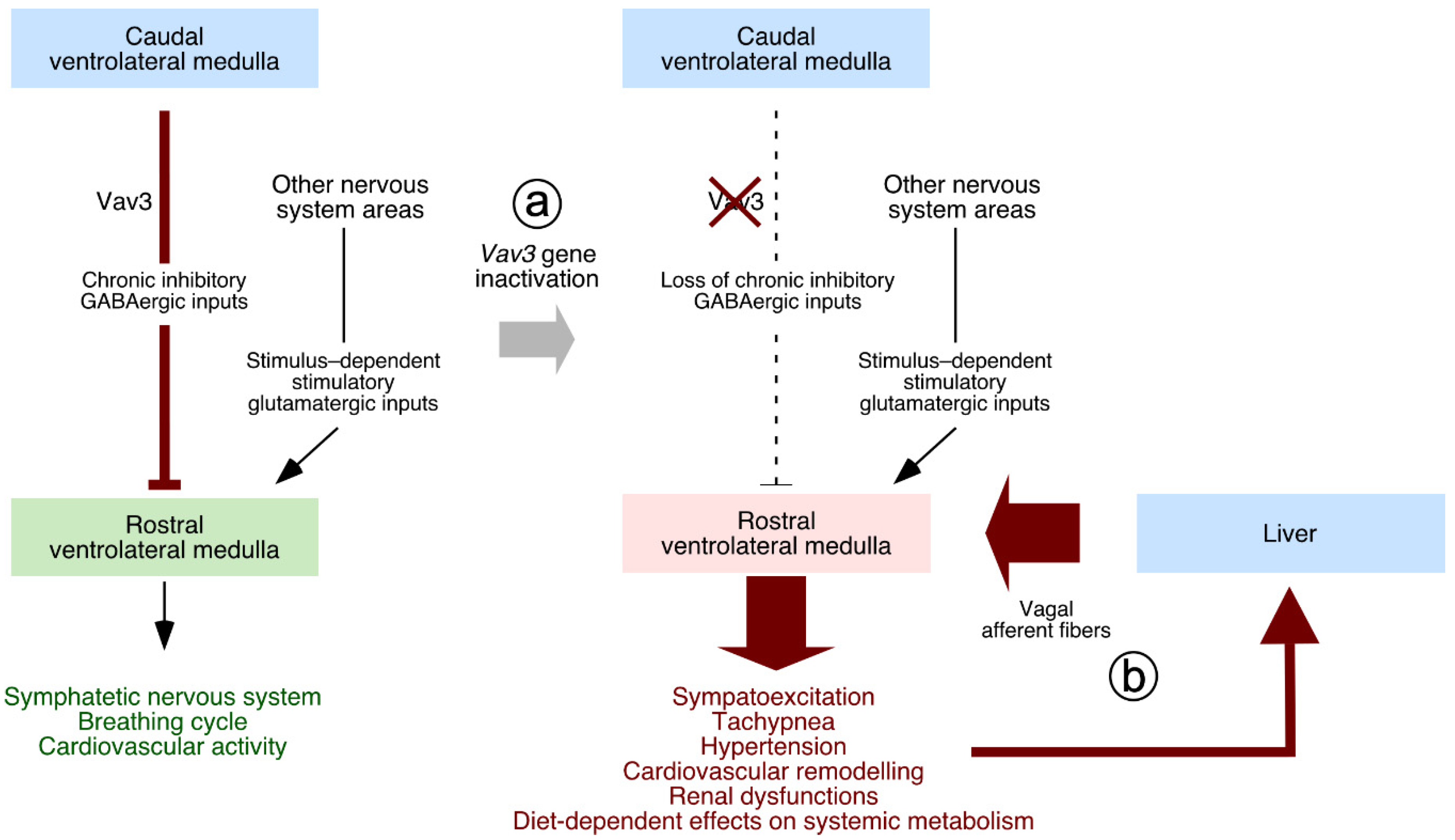
5. Neuron-Associated Vav3 Functions in the Brainstem, Cerebellum, and Retina
6. Vav3, Oligodendrocytes, and Myelination Processes
7. Lessons Learnt from the Phenotypes of Vav Family Knock-Out and Knock-In Mice
8. Physiological Functions of Vav Proteins, a Problem for Potential Anti-Vav Therapies?
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodriguez-Fdez, S.; Bustelo, X.R. The Vav GEF family: An evolutionary and functional perspective. Cells 2019, 8, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzav, S.; Martin-Zanca, D.; Barbacid, M. vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J. 1989, 8, 2283–2290. [Google Scholar] [CrossRef]
- Schuebel, K.E.; Bustelo, X.R.; Nielsen, D.A.; Song, B.J.; Barbacid, M.; Goldman, D.; Lee, I.J. Isolation and characterization of murine vav2, a member of the vav family of proto-oncogenes. Oncogene 1996, 13, 363–371. [Google Scholar] [PubMed]
- Movilla, N.; Bustelo, X.R. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell Biol. 1999, 19, 7870–7885. [Google Scholar] [CrossRef] [Green Version]
- Henske, E.P.; Short, M.P.; Jozwiak, S.; Bovey, C.M.; Ramlakhan, S.; Haines, J.L.; Kwiatkowski, D.J. Identification of VAV2 on 9q34 and its exclusion as the tuberous sclerosis gene TSC1. Ann. Hum. Genet. 1995, 59, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Zugaza, J.L.; Lopez-Lago, M.A.; Caloca, M.J.; Dosil, M.; Movilla, N.; Bustelo, X.R. Structural determinants for the biological activity of Vav proteins. J. Biol. Chem. 2002, 277, 45377–45392. [Google Scholar] [CrossRef] [Green Version]
- Rapley, J.; Tybulewicz, V.L.; Rittinger, K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep. 2008, 9, 655–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrencik, J.E.; Brooun, A.; Zhang, H.; Mathews, I.I.; Hura, G.L.; Foster, S.A.; Perry, J.J.; Streiff, M.; Ramage, P.; Widmer, H.; et al. Structural basis of guanine nucleotide exchange mediated by the T-cell essential Vav1. J. Mol. Biol. 2008, 380, 828–843. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Katzav, S.; Weiss, A. A functional T-cell receptor signaling pathway is required for p95vav activity. Mol. Cell Biol. 1995, 15, 4337–4346. [Google Scholar] [CrossRef] [Green Version]
- Kuhne, M.R.; Ku, G.; Weiss, A. A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J. Biol. Chem. 2000, 275, 2185–2190. [Google Scholar] [CrossRef] [Green Version]
- Doody, G.M.; Billadeau, D.D.; Clayton, E.; Hutchings, A.; Berland, R.; McAdam, S.; Leibson, P.J.; Turner, M. Vav-2 controls NFAT-dependent transcription in B- but not T-lymphocytes. EMBO J. 2000, 19, 6173–6184. [Google Scholar] [CrossRef] [Green Version]
- Robles-Valero, J.; Lorenzo-Martin, L.F.; Menacho-Marquez, M.; Fernandez-Pisonero, I.; Abad, A.; Camos, M.; Toribio, M.L.; Espinosa, L.; Bigas, A.; Bustelo, X.R. A paradoxical tumor-suppressor role for the Rac1 exchange factor Vav1 in T cell acute lymphoblastic leukemia. Cancer Cell 2017, 32, 608–623.e9. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Martins, I.R.; Li, P.; Amarasinghe, G.K.; Umetani, J.; Fernandez-Zapico, M.E.; Billadeau, D.D.; Machius, M.; Tomchick, D.R.; Rosen, M.K. Structural and energetic mechanisms of cooperative autoinhibition and activation of Vav1. Cell 2010, 140, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Fdez, S.; Citterio, C.; Lorenzo-Martin, L.F.; Baltanas-Copado, J.; Llorente-Gonzalez, C.; Corbalan-Garcia, S.; Vicente-Manzanares, M.; Bustelo, X.R. Phosphatidylinositol Monophosphates Regulate Optimal Vav1 Signaling Output. Cells 2019, 8, 1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Fdez, S.; Fernández-Nevado, L.; Lorenzo-Martín, L.F.; Bustelo, X.R. Lysine Acetylation Reshapes the Downstream Signaling Landscape of Vav1 in Lymphocytes. Cells 2020, 9, 609. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Pitari, G.M.; Schulz, S.; Waldman, S.A. Nitric oxide signaling: Systems integration of oxygen balance in defense of cell integrity. Curr. Opin. Hematol. 2004, 11, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Nitric oxide as a unique signaling molecule in the vascular system: A historical overview. J. Physiol. Pharmacol. 2002, 53, 503–514. [Google Scholar] [PubMed]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 2000, 52, 375–414. [Google Scholar] [PubMed]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, D.A.; Takimoto, E.; Nagayama, T.; Champion, H.C. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc. Res. 2007, 75, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, P.A.; Garvin, J.L. Cardiovascular and renal control in NOS-deficient mouse models. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2003, 284, R628–R638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiss, C.; Wang, G.X.; Korth, M.; Aszodi, A.; Andersson, K.E.; et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998, 17, 3045–3051. [Google Scholar] [CrossRef]
- Vallance, P.; Collier, J.; Moncada, S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 1989, 2, 997–1000. [Google Scholar] [CrossRef]
- Ravipati, G.; McClung, J.A.; Aronow, W.S.; Peterson, S.J.; Frishman, W.H. Type 5 phosphodiesterase inhibitors in the treatment of erectile dysfunction and cardiovascular disease. Cardiol. Rev. 2007, 15, 76–86. [Google Scholar] [CrossRef]
- Napoli, C.; Ignarro, L.J. Nitric oxide-releasing drugs. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 97–123. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Jerkic, M.; Lopez-Novoa, J.M.; Bustelo, X.R. Loss of Vav2 proto-oncogene causes tachycardia and cardiovascular disease in mice. Mol. Biol. Cell 2007, 18, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Sauzeau, V.; Sevilla, M.A.; Montero, M.J.; Bustelo, X.R. The Rho/Rac exchange factor Vav2 controls nitric oxide-dependent responses in mouse vascular smooth muscle cells. J. Clin. Investig. 2010, 120, 315–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef]
- Fabbiano, S.; Menacho-Marquez, M.; Sevilla, M.A.; Albarran-Juarez, J.; Zheng, Y.; Offermanns, S.; Montero, M.J.; Bustelo, X.R. Genetic dissection of the Vav2-Rac1 signaling axis in vascular smooth muscle cells. Mol. Cell Biol. 2014, 34, 4404–4419. [Google Scholar] [CrossRef] [Green Version]
- Andre, G.; Sandoval, J.E.; Retailleau, K.; Loufrani, L.; Toumaniantz, G.; Offermanns, S.; Rolli-Derkinderen, M.; Loirand, G.; Sauzeau, V. Smooth muscle specific Rac1 deficiency induces hypertension by preventing p116RIP3-dependent RhoA inhibition. J. Am. Heart Assoc. 2014, 3, e000852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo-Martín, L.F.; Rodríguez-Fdez, S.; Fabbiano, S.; Abad, A.; García-Macías, M.C.; Dosil, M.; Cuadrado, M.; Robles-Valero, J.; Bustelo, X.R. Vav2 pharmaco-mimetic mice reveal the therapeutic value and caveats of the catalytic inactivation of a Rho exchange factor. Oncogene 2020, 39, 5098–5111. [Google Scholar] [CrossRef]
- Rodríguez-Fdez, S.; Lorenzo-Martín, L.F.; Fernández-Pisonero, I.; Porteiro, B.; Veyrat-Durebex, C.; Beiroa, D.; Al-Massadi, O.; Abad, A.; Diéguez, C.; Coppari, R.; et al. Vav2 catalysis-dependent pathways contribute to skeletal muscle growth and metabolic homeostasis. Nat. Commun. 2020, 11, 5808. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef]
- Petersen, K.F.; Shulman, G.I. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 11G–18G. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Physical activity and muscle-brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Christoffolete, M.A.; Silva, W.J.; Ramos, G.V.; Bento, M.R.; Costa, M.O.; Ribeiro, M.O.; Okamoto, M.M.; Lohmann, T.H.; Machado, U.F.; Musaro, A.; et al. Muscle IGF-1-induced skeletal muscle hypertrophy evokes higher insulin sensitivity and carbohydrate use as preferential energy substrate. Biomed. Res. Int. 2015, 2015, 282984. [Google Scholar] [CrossRef]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006, 3, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Michael, M.D.; Previs, S.F.; Peroni, O.D.; Mauvais-Jarvis, F.; Neschen, S.; Kahn, B.B.; Kahn, C.R.; Shulman, G.I. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J. Clin. Investig. 2000, 105, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Camporez, J.P.; Petersen, M.C.; Abudukadier, A.; Moreira, G.V.; Jurczak, M.J.; Friedman, G.; Haqq, C.M.; Petersen, K.F.; Shulman, G.I. Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc. Natl. Acad. Sci. USA 2016, 113, 2212–2217. [Google Scholar] [CrossRef] [Green Version]
- Bruning, J.C.; Michael, M.D.; Winnay, J.N.; Hayashi, T.; Horsch, D.; Accili, D.; Goodyear, L.J.; Kahn, C.R. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 1998, 2, 559–569. [Google Scholar] [CrossRef]
- Hamrick, M.W.; Pennington, C.; Webb, C.N.; Isales, C.M. Resistance to body fat gain in ‘double-muscled’ mice fed a high-fat diet. Int. J. Obes. 2006, 30, 868–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, C.W.; Shao, Y.R.; Sahin, M.; Shamah, S.M.; Lin, M.Z.; Greer, P.L.; Gao, S.; Griffith, E.C.; Brugge, J.S.; Greenberg, M.E. Vav family GEFs link activated Ephs to endocytosis and axon guidance. Neuron 2005, 46, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Zhao, C.; Wu, Y.; Yang, Q.; Shao, A.; Wang, T.; Wu, J.; Yin, Y.; Li, Y.; Hou, J.; et al. Identification of a Vav2-dependent mechanism for GDNF/Ret control of mesolimbic DAT trafficking. Nat. Neurosci. 2015, 18, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Scofield, M.D.; Heinsbroek, J.A.; Gipson, C.D.; Kupchik, Y.M.; Spencer, S.; Smith, A.C.; Roberts-Wolfe, D.; Kalivas, P.W. The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol. Rev. 2016, 68, 816–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauzeau, V.; Horta-Junior, J.A.; Riolobos, A.S.; Fernandez, G.; Sevilla, M.A.; Lopez, D.E.; Montero, M.J.; Rico, B.; Bustelo, X.R. Vav3 is involved in GABAergic axon guidance events important for the proper function of brainstem neurons controlling cardiovascular, respiratory, and renal parameters. Mol. Biol. Cell 2010, 21, 4251–4263. [Google Scholar] [CrossRef] [Green Version]
- Guyenet, P.G. The sympathetic control of blood pressure. Nat. Rev. Neurosci. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Schreihofer, A.M.; Guyenet, P.G. The baroreflex and beyond: Control of sympathetic vasomotor tone by GABAergic neurons in the ventrolateral medulla. Clin. Exp. Pharmacol. Physiol. 2002, 29, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Schreihofer, A.M.; Guyenet, P.G. Baro-activated neurons with pulse-modulated activity in the rat caudal ventrolateral medulla express GAD67 mRNA. J. Neurophysiol. 2003, 89, 1265–1277. [Google Scholar] [CrossRef]
- Cravo, S.L.; Morrison, S.F. The caudal ventrolateral medulla is a source of tonic sympathoinhibition. Brain Res. 1993, 621, 133–136. [Google Scholar] [CrossRef]
- Guyenet, P.G.; Filtz, T.M.; Donaldson, S.R. Role of excitatory amino acids in rat vagal and sympathetic baroreflexes. Brain Res. 1987, 407, 272–284. [Google Scholar] [CrossRef]
- Sauzeau, V.; Sevilla, M.A.; Rivas-Elena, J.V.; de Alava, E.; Montero, M.J.; Lopez-Novoa, J.M.; Bustelo, X.R. Vav3 proto-oncogene deficiency leads to sympathetic hyperactivity and cardiovascular dysfunction. Nat. Med. 2006, 12, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Sauzeau, V.; Carvajal-Gonzalez, J.M.; Riolobos, A.S.; Sevilla, M.A.; Menacho-Marquez, M.; Roman, A.C.; Abad, A.; Montero, M.J.; Fernandez-Salguero, P.; Bustelo, X.R. Transcriptional factor aryl hydrocarbon receptor (Ahr) controls cardiovascular and respiratory functions by regulating the expression of the Vav3 proto-oncogene. J. Biol. Chem. 2011, 286, 2896–2909. [Google Scholar] [CrossRef] [Green Version]
- Carvajal-Gonzalez, J.M.; Mulero-Navarro, S.; Roman, A.C.; Sauzeau, V.; Merino, J.M.; Bustelo, X.R.; Fernandez-Salguero, P.M. The dioxin receptor regulates the constitutive expression of the vav3 proto-oncogene and modulates cell shape and adhesion. Mol. Biol. Cell 2009, 20, 1715–1727. [Google Scholar] [CrossRef] [Green Version]
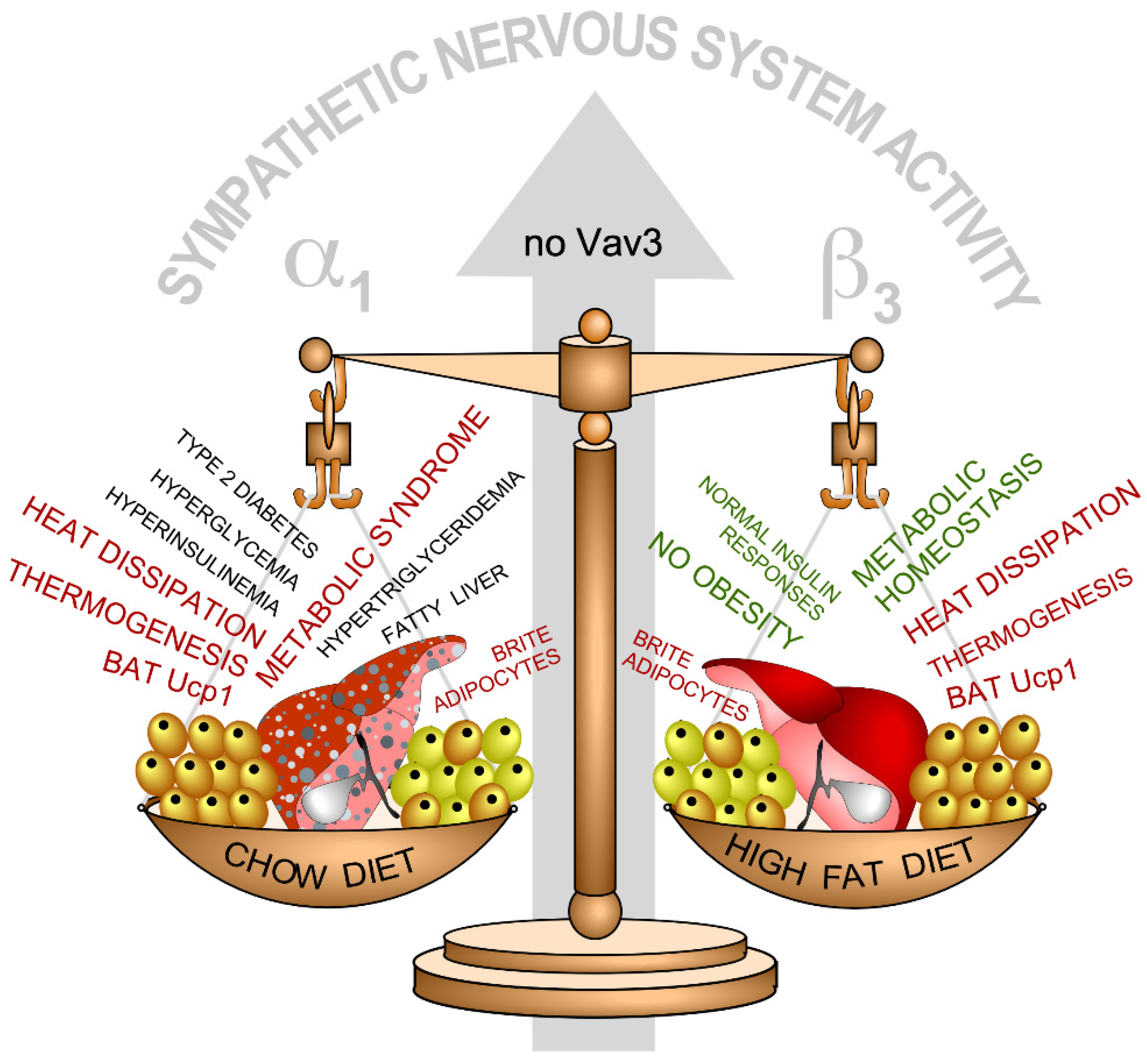
- Menacho-Marquez, M.; Nogueiras, R.; Fabbiano, S.; Sauzeau, V.; Al-Massadi, O.; Dieguez, C.; Bustelo, X.R. Chronic sympathoexcitation through loss of Vav3, a Rac1 activator, results in divergent effects on metabolic syndrome and obesity depending on diet. Cell Metab. 2013, 18, 199–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quevedo, C.; Sauzeau, V.; Menacho-Marquez, M.; Castro-Castro, A.; Bustelo, X.R. Vav3-deficient mice exhibit a transient delay in cerebellar development. Mol. Biol. Cell 2010, 21, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Segal, R.A.; Pomeroy, S.L.; Stiles, C.D. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. J. Neurosci. 1995, 15, 4970–4981. [Google Scholar] [CrossRef]
- Schwartz, P.M.; Borghesani, P.R.; Levy, R.L.; Pomeroy, S.L.; Segal, R.A. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron 1997, 19, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Sadakata, T.; Mizoguchi, A.; Sato, Y.; Katoh-Semba, R.; Fukuda, M.; Mikoshiba, K.; Furuichi, T. The secretory granule-associated protein CAPS2 regulates neurotrophin release and cell survival. J. Neurosci. 2004, 24, 43–52. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, C.J.; Barzik, M.; Fujiwara, I.; Remmert, K.; Wang, Y.X.; Petralia, R.S.; Friedman, T.B.; Hammer, J.A. Myosin 18Aα targets the guanine nucleotide exchange factor β-Pix to the dendritic spines of cerebellar Purkinje neurons and promotes spine maturation. FASEB J. 2021, 35, e21092. [Google Scholar] [CrossRef]
- Tao, T.; Sun, J.; Peng, Y.; Li, Y.; Wang, P.; Chen, X.; Zhao, W.; Zheng, Y.Y.; Wei, L.; Wang, W.; et al. Golgi-resident TRIO regulates membrane trafficking during neurite outgrowth. J. Biol. Chem. 2019, 294, 10954–10968. [Google Scholar] [CrossRef] [PubMed]
- Jaudon, F.; Raynaud, F.; Wehrlé, R.; Bellanger, J.M.; Doulazmi, M.; Vodjdani, G.; Gasman, S.; Fagni, L.; Dusart, I.; Debant, A.; et al. The RhoGEF DOCK10 is essential for dendritic spine morphogenesis. Mol. Biol. Cell 2015, 26, 2112–2127. [Google Scholar] [CrossRef]
- Peng, Y.J.; He, W.Q.; Tang, J.; Tao, T.; Chen, C.; Gao, Y.Q.; Zhang, W.C.; He, X.Y.; Dai, Y.Y.; Zhu, N.C.; et al. Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J. Biol. Chem. 2010, 285, 24834–24844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boverhof, D.R.; Burgoon, L.D.; Tashiro, C.; Chittim, B.; Harkema, J.R.; Jump, D.B.; Zacharewski, T.R. Temporal and dose-dependent hepatic gene expression patterns in mice provide new insights into TCDD-Mediated hepatotoxicity. Toxicol. Sci. 2005, 85, 1048–1063. [Google Scholar] [CrossRef] [Green Version]
- Luft, V.; Reinhard, J.; Shibuya, M.; Fischer, K.D.; Faissner, A. The guanine nucleotide exchange factor Vav3 regulates differentiation of progenitor cells in the developing mouse retina. Cell Tissue Res. 2015, 359, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Ulc, A.; Zeug, A.; Bauch, J.; van Leeuwen, S.; Kuhlmann, T.; Ffrench-Constant, C.; Ponimaskin, E.; Faissner, A. The guanine nucleotide exchange factor Vav3 modulates oligodendrocyte precursor differentiation and supports remyelination in white matter lesions. Glia 2019, 67, 376–392. [Google Scholar] [CrossRef]
- Lorenzo-Martin, L.F.; Menacho-Marquez, M.; Fabbiano, S.; Al-Massadi, O.; Abad, A.; Rodriguez-Fdez, S.; Sevilla, M.A.; Montero, M.J.; Dieguez, C.; Nogueiras, R.; et al. Vagal afferents contribute to sympathoexcitation-driven metabolic dysfunctions. J. Endocrinol. 2019, 240, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Perretta-Tejedor, N.; Fernandez-Mateos, J.; Garcia-Ortiz, L.; Gomez-Marcos, M.A.; Recio-Rodriguez, J.I.; Agudo-Conde, C.; Rodriguez-Sanchez, E.; Morales, A.I.; Lopez-Hernandez, F.J.; Lopez-Novoa, J.M.; et al. Association of VAV2 and VAV3 polymorphisms with cardiovascular risk factors. Sci. Rep. 2017, 7, 41875. [Google Scholar] [CrossRef] [PubMed]
- Miramontes-González, J.P.; Usategui-Martín, R.; Martín-Vallejo, J.; Ziegler, M.; de Isla, L.L.; Connor, D.O.; González-Sarmiento, R. VAV3 rs7528153 and VAV3-AS1 rs1185222 polymorphisms are associated with an increased risk of developing hypertension. Eur. J. Intern. Med. 2020, 80, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef]
- Menacho-Marquez, M.; Garcia-Escudero, R.; Ojeda, V.; Abad, A.; Delgado, P.; Costa, C.; Ruiz, S.; Alarcon, B.; Paramio, J.M.; Bustelo, X.R. The Rho exchange factors Vav2 and Vav3 favor skin tumor initiation and promotion by engaging extracellular signaling loops. PLoS Biol. 2013, 11, e1001615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo-Martín, L.F.; Fernández-Parejo, N.; Menacho-Márquez, M.; Rodríguez-Fdez, S.; Robles-Valero, J.; Zumalave, S.; Fabbiano, S.; Pascual, G.; García-Pedrero, J.M.; Abad, A.; et al. VAV2 signaling promotes regenerative proliferation in both cutaneous and head and neck squamous cell carcinoma. Nat. Commun. 2020, 11, 4788. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Sanchez-Aguilera, A.; Shen, S.; Sengupta, A.; Madhu, M.N.; Ficker, A.M.; Dunn, S.K.; Kuenzi, A.M.; Arnett, J.L.; Santho, R.A.; et al. Vav3 collaborates with p190-BCR-ABL in lymphoid progenitor leukemogenesis, proliferation, and survival. Blood 2012, 120, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Fdez, S.; Lorenzo-Martín, L.F.; Fabbiano, S.; Menacho-Márquez, M.; Sauzeau, V.; Dosil, M.; Bustelo, X.R. New Functions of Vav Family Proteins in Cardiovascular Biology, Skeletal Muscle, and the Nervous System. Biology 2021, 10, 857. https://doi.org/10.3390/biology10090857
Rodríguez-Fdez S, Lorenzo-Martín LF, Fabbiano S, Menacho-Márquez M, Sauzeau V, Dosil M, Bustelo XR. New Functions of Vav Family Proteins in Cardiovascular Biology, Skeletal Muscle, and the Nervous System. Biology. 2021; 10(9):857. https://doi.org/10.3390/biology10090857
Chicago/Turabian StyleRodríguez-Fdez, Sonia, L. Francisco Lorenzo-Martín, Salvatore Fabbiano, Mauricio Menacho-Márquez, Vincent Sauzeau, Mercedes Dosil, and Xosé R. Bustelo. 2021. "New Functions of Vav Family Proteins in Cardiovascular Biology, Skeletal Muscle, and the Nervous System" Biology 10, no. 9: 857. https://doi.org/10.3390/biology10090857
APA StyleRodríguez-Fdez, S., Lorenzo-Martín, L. F., Fabbiano, S., Menacho-Márquez, M., Sauzeau, V., Dosil, M., & Bustelo, X. R. (2021). New Functions of Vav Family Proteins in Cardiovascular Biology, Skeletal Muscle, and the Nervous System. Biology, 10(9), 857. https://doi.org/10.3390/biology10090857