
Metabolic Regulation of Sugar Assimilation for Lipid Production in Aspergillus oryzae BCC7051 through Comparative Transcriptome Perspective
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Cultivation Conditions
2.2. Growth and Residual Sugar Measurements
2.3. Fatty Acid and Lipid Analysis
2.4. RNA Extraction and Sequencing
2.5. RNA Sequence Data Analysis
2.6. Transcriptome Annotation and Pathway Analysis
3. Results
3.1. Growth Kinetic and Lipid Production of A. oryzae BCC7051 on Different Sugars
3.2. Genome-Wide Transcriptome Data of A. oryzae BCC7051
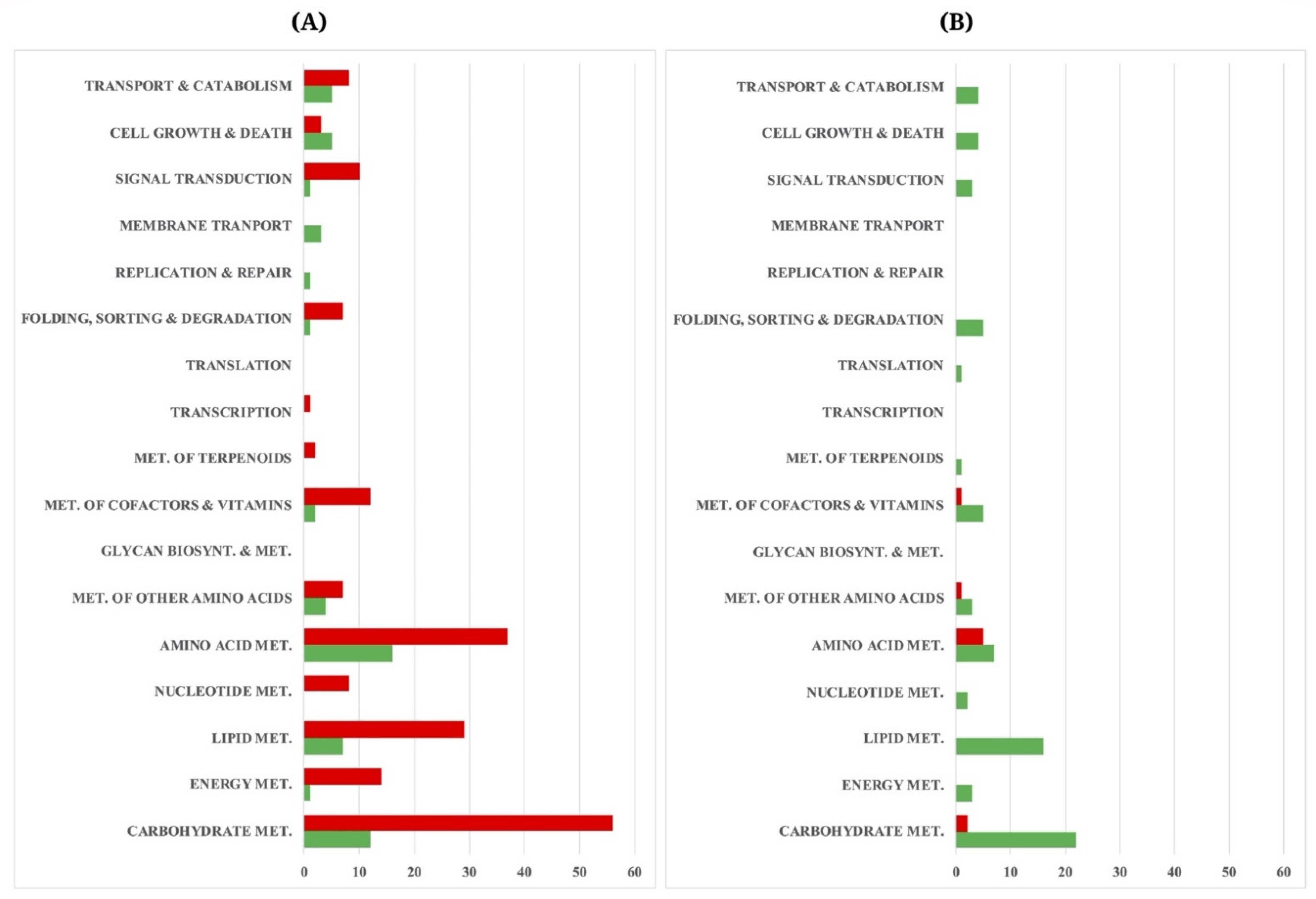
3.3. Differential Gene Expression and Functional Assignment of the A. oryzae Cultures Using Different Carbon Sources
3.4. Metabolic Network of Xylose Assimilation for A. oryzae Growth through Comparative Transcriptome Analysis
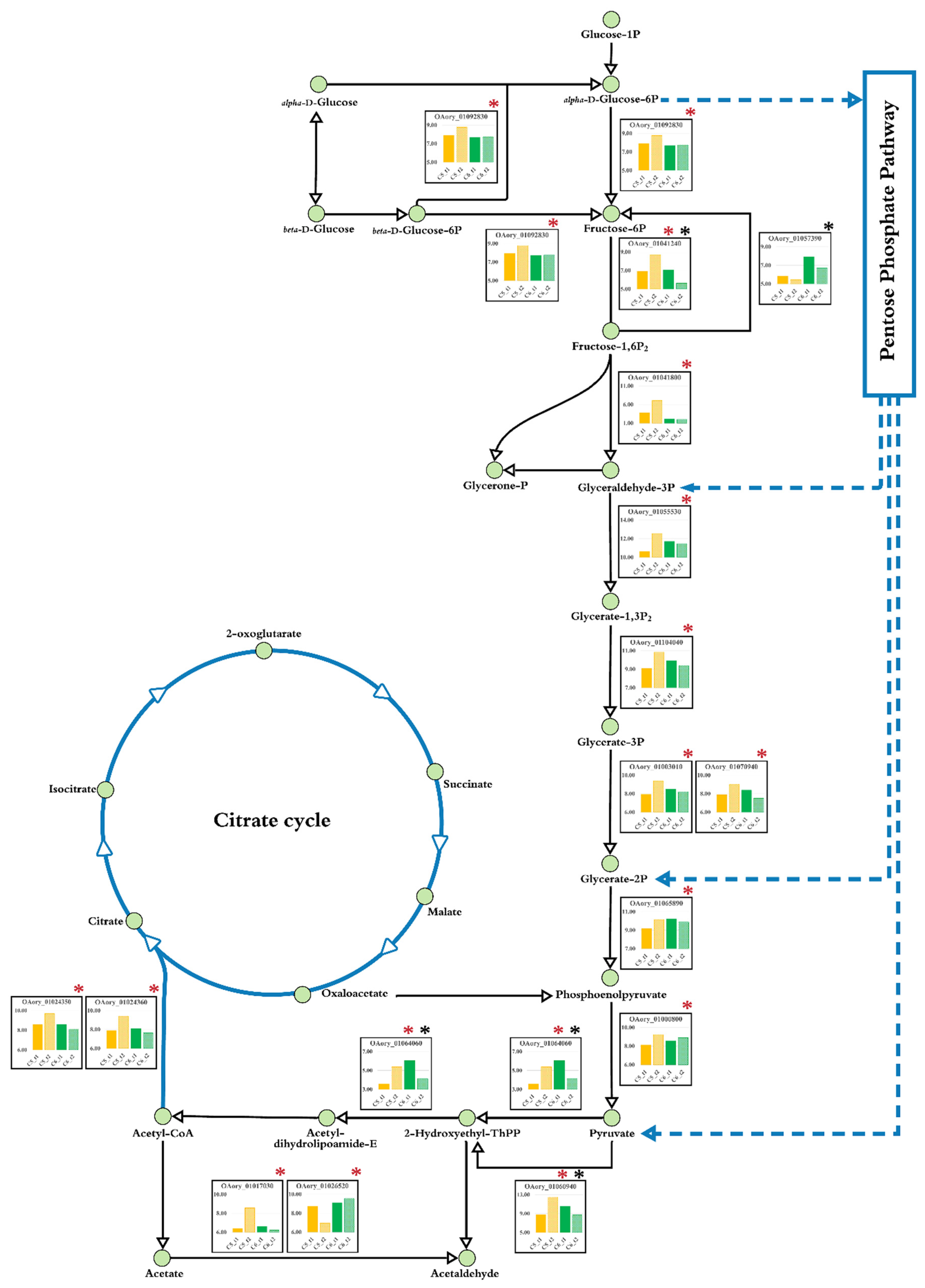
3.5. Metabolic Network of Glucose Assimilation for A. oryzae Growth through Comparative Transcriptome Analysis
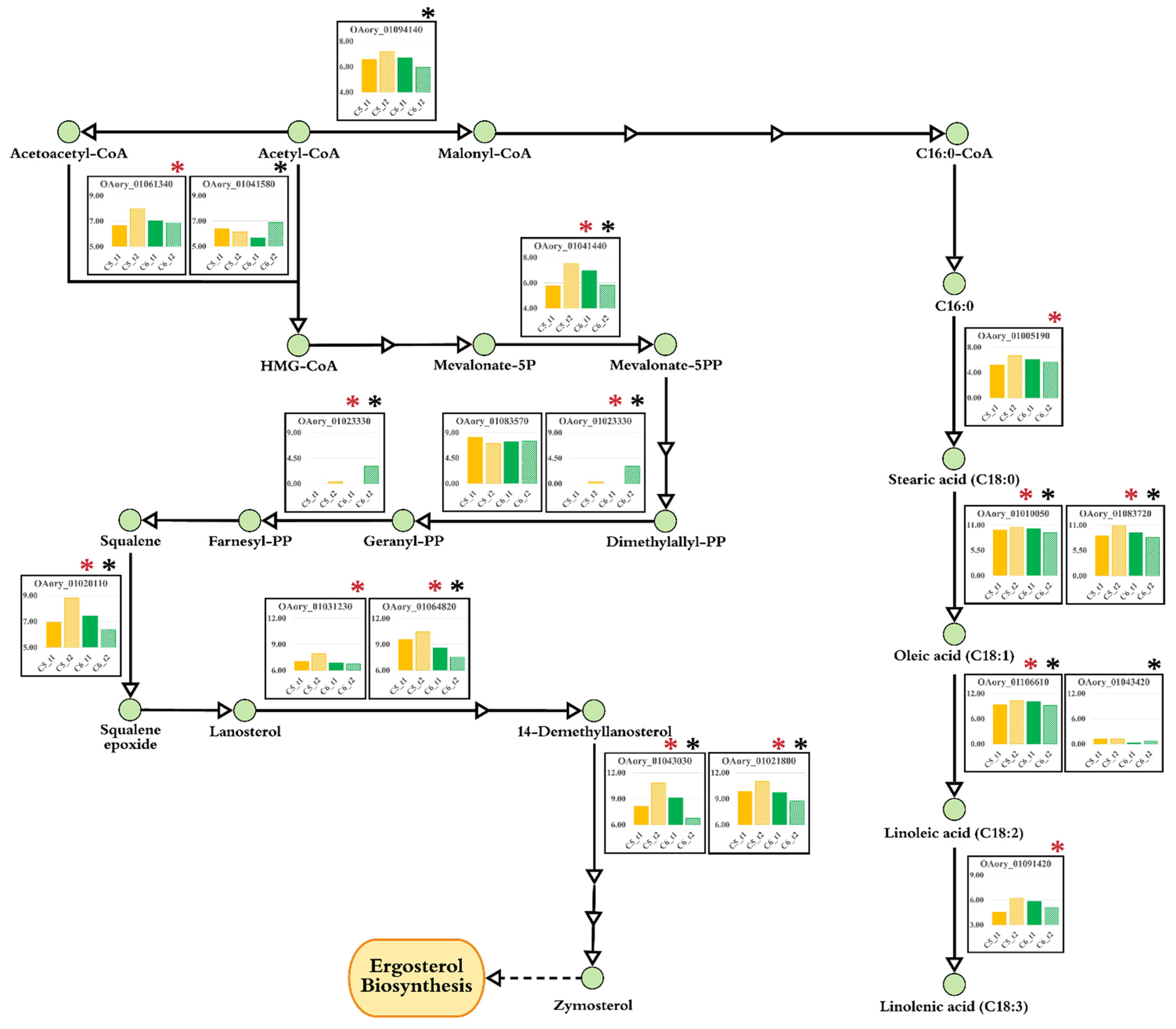
3.6. Transcriptional Alterations at Lipid Accumulation Phases of A. oryzae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyer, V.; Basenko, E.Y.; Benz, J.P.; Braus, G.H.; Caddick, M.X.; Csukai, M.; de Vries, R.P.; Endy, D.; Frisvad, J.C.; Gunde-Cimerman, N.; et al. Growing a circular economy with fungal biotechnology: A white paper. Fungal. Biol. Biotechnol. 2020, 7, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, M.; Lee, Y.H.; Hayashi, R.; Suzuki, Y.; Yamada, O.; Sakamoto, K.; Gotoh, K.; Akita, O. Molecular analysis of an inactive aflatoxin biosynthesis gene cluster in Aspergillus oryzae RIB strains. Appl. Environ. Microbiol. 2006, 72, 484. [Google Scholar] [CrossRef] [Green Version]
- Thammarongtham, C.; Nookaew, I.; Vorapreeda, T.; Srisuk, T.; Land, M.L.; Jeennor, S.; Laoteng, K. Genome characterization of oleaginous Aspergillus oryzae BCC7051: A potential fungal-based platform for lipid production. Curr. Microbiol. 2018, 75, 57–70. [Google Scholar] [CrossRef]
- He, B.; Tu, Y.; Jiang, C.; Zhang, Z.; Li, Y.; Zeng, B. Functional genomics of Aspergillus oryzae: Strategies and progress. Microorganisms 2019, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Hu, Z.; Ma, L.; Li, H.; Ai, M.; Han, J.; Zeng, B. Transcriptome analysis of different growth stages of Aspergillus oryzae reveals dynamic changes of distinct classes of genes during growth. BMC Microbiol. 2018, 18, 12. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Guo, G.; Wang, C.; Lin, Y.; Wang, X.; Zhao, M.; Guo, Y.; He, M.; Zhang, Y.; Pan, L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acid. Res. 2010, 38, 5075–5087. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Yao, Y.; Wang, C.; Tian, F.; Liu, X.; Hou, L.; Yang, Z.; Zhao, J.; Zhang, H.; Cao, X. Transcriptome and proteome expression analysis of the metabolism of amino acids by the fungus Aspergillus oryzae in fermented soy sauce. BioMed Res. Intern. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Antimanon, S.; Anantayanon, J.; Wannawilai, S.; Khongto, B.; Laoteng, K. Physiological traits of dihomo-γ-linolenic acid production of the engineered Aspergillus oryzae by comparing mathematic models. Front. Microbiol. 2020, 11, 546230. [Google Scholar] [CrossRef]
- Meng, X.; Yang, J.; Xu, X.; Zhang, L.; Nie, Q.; Xian, M. Biodiesel production from oleaginous microorganisms. Renew. Energy 2009, 34, 1–5. [Google Scholar] [CrossRef]
- Chutrakul, C.; Jeennor, S.; Panchanawaporn, S.; Cheawchanlertfa, P.; Suttiwattanakul, S.; Mayura, V.; Laoteng, K. Metabolic engineering of long chain-polyunsaturated fatty acid biosynthetic pathway in oleaginous fungus for dihomo-gamma linolenic acid production. J. Biotechnol. 2016, 218, 85–93. [Google Scholar] [CrossRef]
- Jeennor, S.; Anantayanon, J.; Panchanawaporn, S.; Khoomrung, S.; Chutrakul, C.; Laoteng, K. Reengineering Aspergillus oryzae to enhance dihomo-gamma linolenic acid production using integrative approach. Gene 2019, 706, 106–114. [Google Scholar] [CrossRef]
- Vorapreeda, T.; Thammarongtham, C.; Cheevadhanarak, S.; Laoteng, K. Alternative routes of acetyl-CoA synthesis identified by comparative genomic analysis: Involvement in the lipid production of oleaginous yeast and fungi. Microbiology 2012, 158, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Wannawilai, S.; Jeennor, S.; Khongto, B.; Laoteng, K. Exploring differential traits of lipid-producing stages of the wild type and morphologically engineered strain of Aspergillus oryzae by comparative kinetic modeling. World J. Microbiol. Biotechnol. 2020, 36, 183. [Google Scholar] [CrossRef]
- Gao, D.; Zeng, J.; Zheng, Y.; Yu, X.; Chen, S. Microbial lipid production from xylose by Mortierella isabellina. Bioresour. Technol. 2013, 133, 315–321. [Google Scholar] [CrossRef]
- Zeng, J.; Zheng, Y.; Yu, X.; Yu, L.; Gao, D.; Chen, S. Lignocellulosic biomass as a carbohydrate source for lipid production by Mortierella isabellina. Bioresour. Technol. 2013, 128, 385–391. [Google Scholar] [CrossRef]
- Díaz-Fernández, D.; Aguiar, T.Q.; Martín, V.I.; Romaní, A.; Silva, R.; DominguesJosé, L.; Revuelta, J.L.; Jiménez, A. Microbial lipids from industrial wastes using xylose-utilizing Ashbya gossypii strains. Bioresour. Technol. 2019, 293, 122054. [Google Scholar] [CrossRef] [PubMed]
- Dourou, M.; Aggeli, D.; Papanikolaou, S.; Aggelis, G. Critical steps in carbon metabolism affecting lipid accumulation and their regulation in oleaginous microorganisms. Appl. Microbiol. Biotechnol. 2018, 102, 2509–2523. [Google Scholar] [CrossRef]
- Gong, Z.; Wang, Q.; Shen, H.; Hu, C.; Jin, G.; Zhao, Z.K. Co-fermentation of cellobiose and xylose by Lipomyces starkeyi for lipid production. Bioresour. Technol. 2012, 117, 20–24. [Google Scholar] [CrossRef]
- Jeennor, S.; Laoteng, K.; Tanticharoen, M.; Cheevadhanarak, S. Evaluation of inoculum performance for enhancing gamma-linolenic acid production from Mucor rouxii. Lett. Appl. Microbiol. 2008, 46, 421–427. [Google Scholar] [CrossRef]
- Lepage, G.; Roy, C.C. Improved recovery of fatty acid through direct transesterification without prior extraction or purification. J. Lipid Res. 1984, 25, 1391–1396. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Khoomrung, S.; Chumnanpuen, P.; Jansa-Ard, S.; Staìšhlman, M.; Nookaew, I.; Borén, J.; Nielsen, J. Rapid quantification of yeast lipid using microwave-assisted total lipid extraction and HPLC-CAD. Anal. Chem. 2013, 85, 4912–4919. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowties. Nat. Methods 2013, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq–A python framework to work with high-throughput sequencing data. Bioinformatics 2015, 15, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelly, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2013, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acid. Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratledge, C.; Wynn, J.P. The biochemistry and molecular biology of lipid accumulation in oleaginous microorganisms. Adv. Appl. Microbiol. 2002, 51, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Laoteng, K.; Certik, M.; Cheevadhanarak, S. Mechanisms controlling lipid accumulation and polyunsaturated fatty acid synthesis in oleaginous fungi. Chem. Pap. 2011, 65, 97–103. [Google Scholar] [CrossRef]
- Laoteng, K.; Jitsue, S.; Dandusitapunth, Y.; Cheevadhanarak, S. Ethanol-induced changes in expression profiles of cell growth, fatty acid and desaturase genes of Mucor rouxii. Fungal Genet. Biol. 2008, 45, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ha, S.J.; Kim, H.J.; Galazka, J.M.; Cate, J.H.; Jin, Y.S.; Zhao, H. Investigation of the functional role of aldose 1-epimerase in engineered cellobiose utilization. J. Biotechnol. 2013, 168, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tewari, Y.B.; Goldberg, R.N. Thermodynamics of reactions catalyzed by L-iditol 2-dehydrogenase: The xylose assimilation pathway. J. Chem. Thermodyn. 1996, 28, 1127–1144. [Google Scholar] [CrossRef]
- Singh, R.P.; Shelke, G.M.; Kumar, A.; Jha, P.N. Biochemistry and genetics of ACC deaminase: A weapon to “stress ethylene” produced in plants. Front. Microbiol. 2015, 6, 937. [Google Scholar] [CrossRef]
- Madi, L.; McBride, S.A.; Bailey, L.A.; Ebbole, D.J. rco-3, a gene involved in glucose transport and conidiation in Neurospora crassa. Genetics 1997, 146, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Q.; Li, J.; Lin, L.; Li, X.; Zhang, Y.; Tian, C. RCO-3 and COL-26 form an external-to-internal module that regulates the dual-affinity glucose transport system in Neurospora crassa. Biotechnol. Biofuels 2021, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Berndt, J.; Boll, M.; Lowel, M.; Gaumert, R. Regulation of sterol biosynthesis in yeast: Induction of 3-hydroxy-3-methylglutaryl-CoA reductase by glucose. Biochem. Biphys. Res. Commun. 1973, 51, 843–848. [Google Scholar] [CrossRef]
- Boll, M.; Lowel, M.; Still, J.; Berndt, J. Sterol biosynthesis in yeast. 3-hydroxy-3-methylglutaryl-coenzyme A reductase as a regulatory enzyme. Eur. J. Biochem. 1975, 54, 435–444. [Google Scholar] [CrossRef]
- Alcazar-Fuoli, L.; Mellado, E. Ergosterol biosynthesis in Aspergillus fumigatus: Its relevance as an antifungal target and role in antifungal drug resistance. Front. Microbiol. 2013, 3, 439. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Esquivel, B.D.; White, T.C. Overexpression or deletion of ergosterol biosynthesis genes alters doubling time, response to stress agents, and drug susceptibility in Saccharomyces cerevisiae. mBio 2018, 9, e01291-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Ma, P.; Li, C.; Xiao, L.; Liang, Z.; Dong, J. Combining transcriptomics and metabolics to reveal the underlying molecular mechanism of ergosterol biosynthesis during the fruiting process of Flammulina veluyipes. BMC Genomics 2019, 20, 999. [Google Scholar] [CrossRef] [Green Version]
- Jardon, R.; Gancedo, C.; Flores, C.L. The gluconeogenic enzyme fructose-1,6-biphosphatase is dispensable for growth of the yeast Yarrowia lipolytica in gluconeogenic substrates. Eukaryot. Cell 2008, 7, 1742–1749. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, A.; Bär, J. Molecular genetics of 6-phosphofructokinase in Pichia pastoris. Yeast 2002, 19, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hoek, P.; Flikweert, M.T.; van der Aart, Q.J.; Steensma, H.Y.; van Dijken, J.P.; Pronk, J.T. Effects of pyruvate decarboxylase overproduction on flux distribution at the pyruvate branch point in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 1998, 64, 2133–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehan, J.; Dunahay, T.; Benemann, J.; Roessler, P.G.; US Department of Energy’s Office of Fuels Development. A look back at the US Department of Energy’s aquatic species program—Biodiesel from algae. In 1998 Close Out Report TP-580-24190; National Renewable Energy Laboratory: Golden, CO, USA, 1998. [Google Scholar]
- Leyva, L.A.; Bashan, Y.; de-Bashan, L.E. Activity of acetyl-CoA carboxylase is not directly linked to accumulation of lipids when Chlorella vulgaris is co-immobilised with Azospirillum brasilense in alginate under autotrophic and heterotrophic conditions. Ann. Microbiol. 2015, 65, 339–349. [Google Scholar] [CrossRef]
- Tamano, K.; Bruno, K.S.; Karagiosis, S.A.; Culley, D.E.; Deng, S.; Collett, J.R.; Umemura, M.; Koike, H.; Baker, S.E.; Machida, M. Increased production of fatty acids and triglycerides in Aspergillus oryzae by enhancing expressions of fatty acid synthesis-related genes. Appl. Microbiol. Biotechnol. 2013, 97, 269–281. [Google Scholar] [CrossRef]
- Chun, K.Y.; Vinarov, D.A.; Zajicek, J.; Miziorko, H.M. 3-Hydroxy-3-methylglutaryl-CoA synthase. A role for glutamate 95 in general acid/base catalysis of C–C bond formation. J. Biol. Chem. 2000, 275, 17946–17953. [Google Scholar] [CrossRef] [Green Version]
- Qiang, S.; Wang, J.; Xiong, X.C.; Qu, Y.L.; Liu, L.; Hu, C.Y.; Meng, Y.H. Promoting the synthesis of precursor substances by overexpressing hexokinase (Hxk) and hydroxymethylglutaryl-CoA synthase (Erg13) to elevate β-carotene production in engineered Yarrowia lipolytica. Front. Mirobiol. 2020, 11, 1346. [Google Scholar] [CrossRef]
- Groppi, S.; Belotti, F.; Brandão, R.L.; Martegani, E.; Tisi, R. Glucose-induced calcium influx in budding yeast involves a novel calcium transport system and can activate calcineurin. Cell Calcium 2011, 49, 376–386. [Google Scholar] [CrossRef]
- Cyert, M.S.; Philpott, C.C. Regulation of cation balance in Saccharomyces cerevisiae. Genetics 2013, 193, 677–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisi, R.; Rigamonti, M.; Groppi, S.; Belotti, F. Calcium homeostasis and signaling in fungi and their relevance for pathogenicity of yeasts and filamentous fungi. AIMS Mol. Sci. 2016, 3, 505–549. [Google Scholar] [CrossRef]
- Berg, J.; Yang, H.; Jan, L.Y. Ca2+-activated Cl− channels at a glance. J. Cell Sci. 2012, 125, 1367–1371. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Sun, J.; Nimtz, M.; Wissing, J.; Zeng, A.P.; Rinas, U. The intra- and extracellular proteome of Aspergillus niger growing on defined medium with xylose or maltose as carbon substrate. Microb. Cell Fact. 2010, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lin, L.; Li, H.; Tian, C.; Ma, Y. Transcriptional comparison of the filamentous fungus Neurospora crassa growing on three major monosaccharides D-glucose, D-xylose and L-arabinose. Biotechnol. Biofuels 2014, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Murakoshi, Y.; Makita, T.; Kato, M.; Kobayashi, T. Comparison and characterization of α-amylase inducers in Aspergillus nidulans based on nuclear localization of AmyR. Appl. Microbiol. Biotechnol. 2012, 94, 1629–1635. [Google Scholar] [CrossRef] [Green Version]
- vanKuyk, P.A.; Benen, J.A.E.; Wösten, H.A.B.; Visser, J.; de Vries, R.P. A broader role for AmyR in Aspergillus niger: Regulation of the utilisation of d-glucose or d-galactose containing oligo- and polysaccharides. Appl. Microbiol. Biotechnol. 2012, 93, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Tanaka, M.; Konno, Y.; Ichikawa, T.; Ichinose, S.; Hasegawa-Shiro, S.; Shintani, T.; Gomi, K. Distinct mechanism of activation of two transcription factors, AmyR and MalR, involved in amylolytic enzyme production in Aspergillus oryzae. Appl. Microbiol. Biotechnol. 2015, 99, 1805–1815. [Google Scholar] [CrossRef]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef]
- Zhang, K.; Huang, B.; Yuan, K.; Ji, X.; Song, P.; Ding, Q.; Wang, Y. Comparative transcriptomics analysis of the responses of the filamentous fungus Glarea lozoyensis to different carbon sources. Front. Microbiol. 2020, 6, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | C5 Cultures | C6 Cultures | ||
|---|---|---|---|---|
| C5_t1 a | C5_t2 b | C6_t1 c | C6_t2 d | |
| DCW (g L−1) | 7.07 ± 0.10 A | 11.66 ± 0.03 C | 7.48 ± 0.15 A | 10.85 ± 0.21 D |
| Lipid content (% in DCW, w/w) | 8.00 ± 2.55 A | 17.70 ± 0.42 C | 8.80 ± 0.28 A | 14.40 ± 0.00 D |
| Residual sugar concentration (g L−1) | 57.80 ± 0.00 A | 22.27 ± 0.08 C | 43.27 ± 0.12 B | 10.77 ± 0.00 D |
| QX (g L−1 day−1) | 4.36 ± 0.07 A | 2.78 ± 0.01 C | 4.63 ± 0.10 A | 3.44 ± 0.07 D |
| QP (g L−1 day−1) | 0.38 ± 0.13 A | 0.52 ± 0.01 C | 0.44 ± 0.02 A | 0.52 ± 0.01 C |
| QS (mol L−1 day−1) | 0.53 ± 0.00 A | 0.49 ± 0.00 C | 0.82 ± 0.00 B | 0.77 ± 0.00 D |
| YX/S (g mol−1) | 8.28 ± 0.13 A | 5.65 ± 0.02 C | 5.65 ± 0.12 B | 4.47 ± 0.00 D |
| YP/S (g mol−1) | 0.72 ± 0.04 A | 1.05 ± 0.03 C | 0.53 ± 0.03 B | 0.68 ± 0.01 D |
| Proportion in Total Lipid (% w/w) | C5 Cultures | C6 Cultures | ||
|---|---|---|---|---|
| C5_t1 a | C5_t2 b | C6_t1 c | C6_t2 d | |
| SE | 12.0 ± 1.1 A | 13.5 ± 1.7 C | 11.6 ± 0.6 A | 9.2 ± 1.6 D |
| TAG | 30.7 ± 0.2 A | 34.9 ± 1.7 C | 30.9 ± 2.6 A | 34.7 ± 4.0 C |
| FFA | 28.2 ± 0.7 A | 23.1 ± 2.6 C | 31.3 ± 1.7 B | 28.9 ± 1.9 D |
| PL | 5.3 ± 0.2 A | 5.6 ± 0.1 C | 5.1 ± 0.4 A | 5.6 ± 0.3 C |
| Others | 23.8 ± 0.8 A | 22.9 ± 2.6 C | 21.1 ± 1.9 A | 21.6 ± 4.1 C |
| Proportion in Total Fatty Acid (% w/w) | C5 Cultures | C6 Cultures | ||
|---|---|---|---|---|
| C5_t1 a | C5_t2 b | C6_t1 c | C6_t2 d | |
| C14:0 | 0.0 ± 0.0 A | 0.2 ± 0.0 C | 0.0 ± 0.0 A | 0.0 ± 0.0 C |
| C16:0 | 15.9 ± 0.6 A | 15.9 ± 0.1 C | 13.4 ± 0.0 B | 14.1 ± 0.0 D |
| C16:1Δ9 | 0.9 ± 0.0 A | 0.4 ± 0.0 C | 0.0 ± 0.0 B | 0.0 ± 0.0 C |
| C18:0 | 13.3 ± 0.1 A | 18.7 ± 0.1 C | 11.6 ± 0.2 B | 16.1 ± 0.4 D |
| C18:1Δ9 | 28.9 ± 0.2 A | 33.5 ± 0.2 C | 25.9 ± 0.1 B | 29.9 ± 0.4 D |
| C18:2Δ9,12 | 40.1 ± 0.3 A | 29.8 ± 0.0 C | 48.5 ± 0.3 B | 39.0 ± 0.7 D |
| C18:3Δ9,12,15 | 0.0 ± 0.0 A | 0.0 ± 0.0 C | 0.0 ± 0.0 A | 0.0 ± 0.0 C |
| C20:0 | 0.9 ± 0.0 A | 1.5 ± 0.0 C | 0.6 ± 0.0 A | 0.9 ± 0.2 D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorapreeda, T.; Khongto, B.; Thammarongtham, C.; Srisuk, T.; Laoteng, K. Metabolic Regulation of Sugar Assimilation for Lipid Production in Aspergillus oryzae BCC7051 through Comparative Transcriptome Perspective. Biology 2021, 10, 885. https://doi.org/10.3390/biology10090885
Vorapreeda T, Khongto B, Thammarongtham C, Srisuk T, Laoteng K. Metabolic Regulation of Sugar Assimilation for Lipid Production in Aspergillus oryzae BCC7051 through Comparative Transcriptome Perspective. Biology. 2021; 10(9):885. https://doi.org/10.3390/biology10090885
Chicago/Turabian StyleVorapreeda, Tayvich, Bhimabol Khongto, Chinae Thammarongtham, Tanawut Srisuk, and Kobkul Laoteng. 2021. "Metabolic Regulation of Sugar Assimilation for Lipid Production in Aspergillus oryzae BCC7051 through Comparative Transcriptome Perspective" Biology 10, no. 9: 885. https://doi.org/10.3390/biology10090885