
Mitochondrial Heterogeneity in Metabolic Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
1.1. What Is Heterogeneity and Why Should We Care?
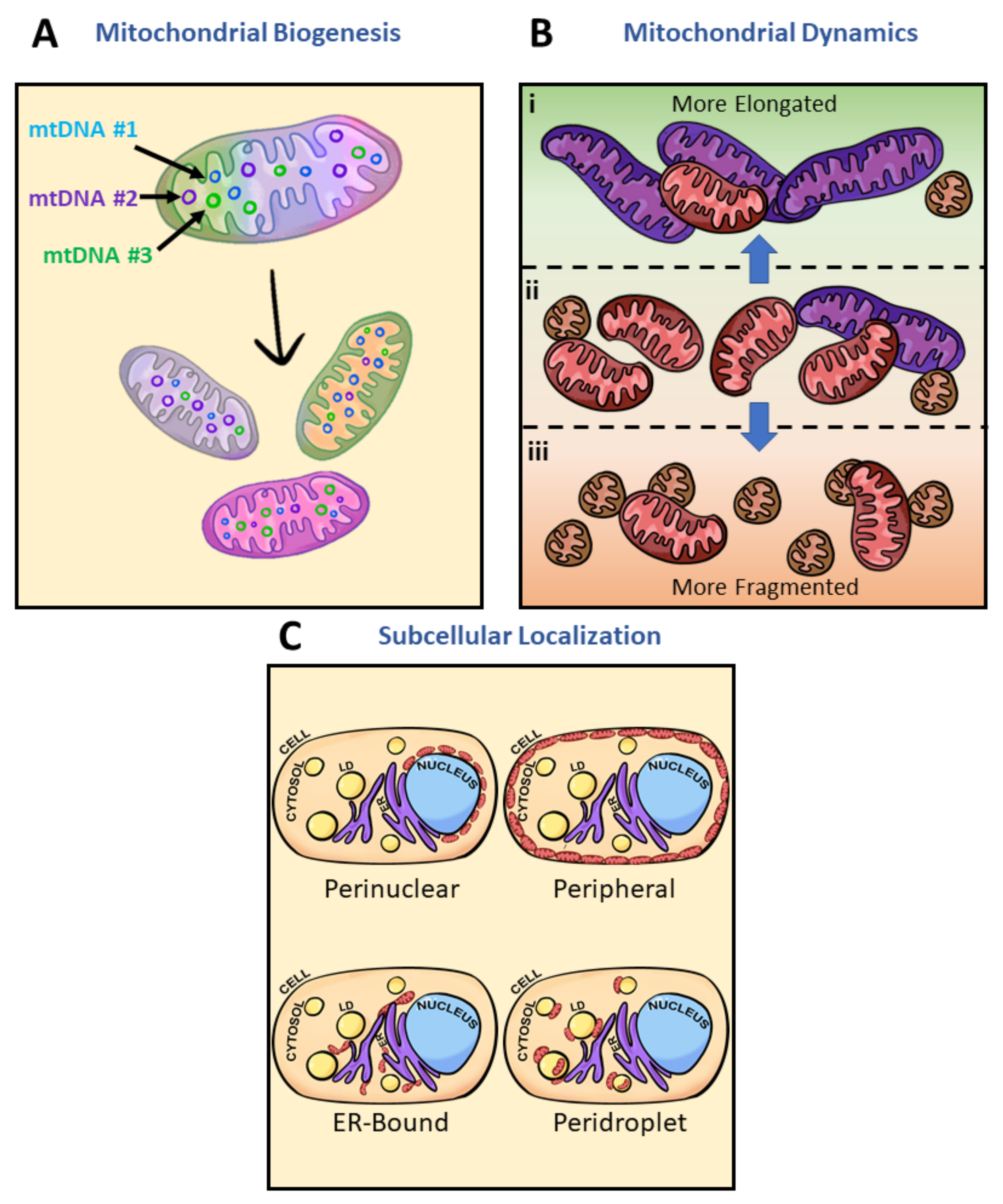
1.2. Nongenetic Contributors to Mitochondrial Homogeneity and Heterogeneity
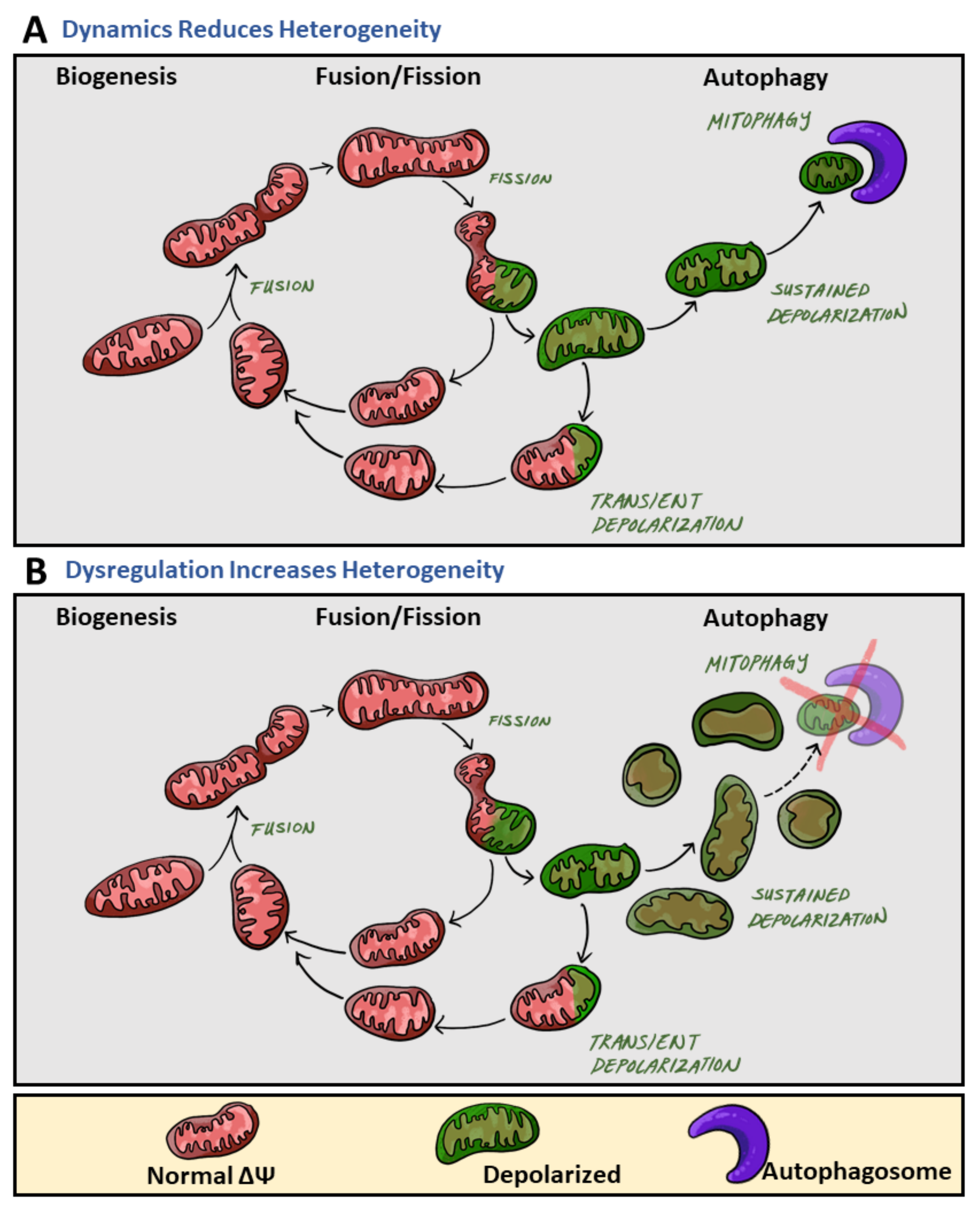
2. Mitochondrial Heterogeneity Increases under Pathological States
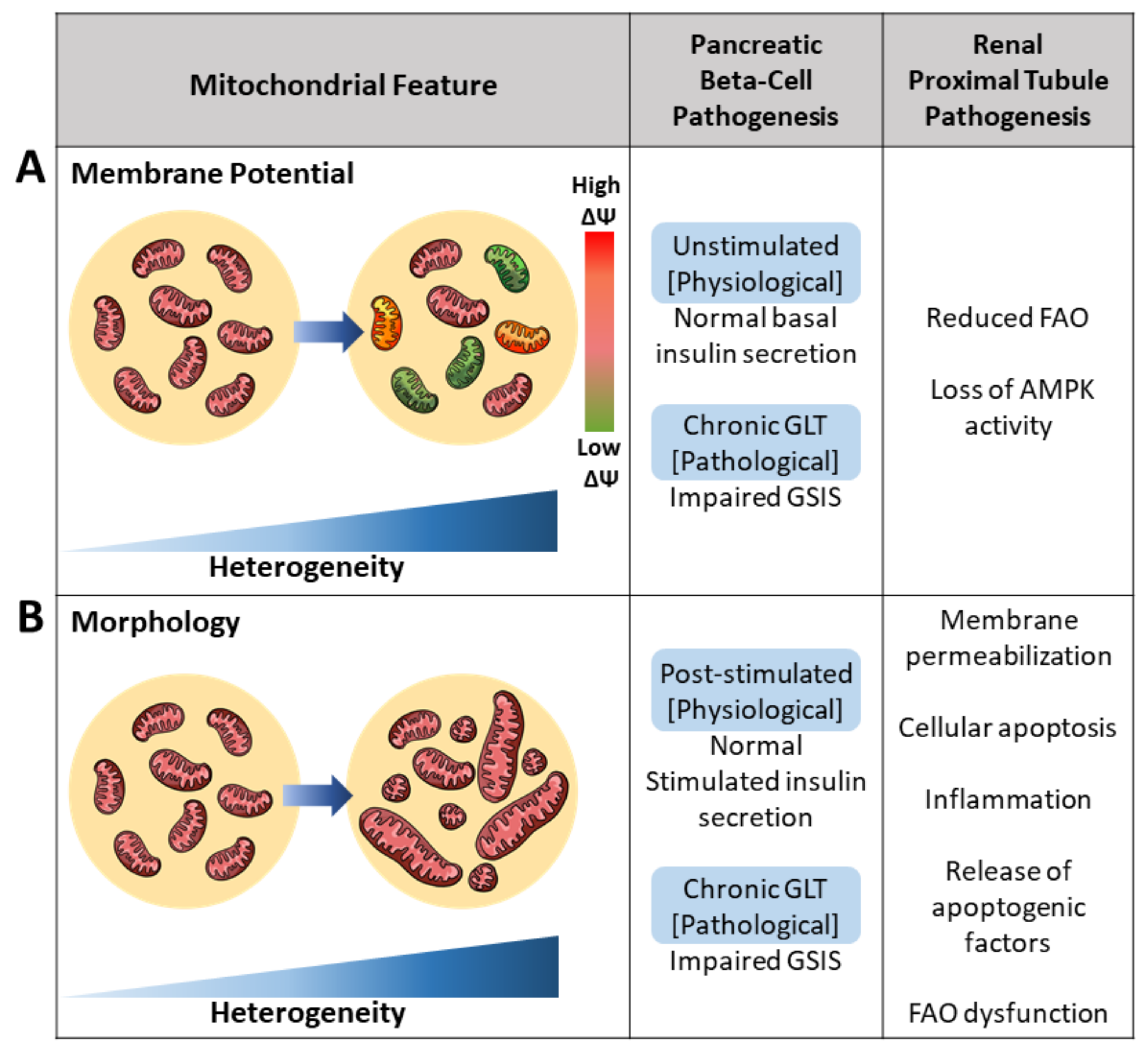
2.1. Membrane Potential Heterogeneity Reflects Diverse Mitochondrial Response to Cellular Nutrient Load
2.2. Brief Overview of Mediators in Mitochondrial Fusion and Fission
2.3. Architectural Variance under Pathological States Introduces Metabolic Defects
2.4. Impaired Mitochondrial Calcium Buffering Drives Pathogenesis in Mitochondrial Signaling
3. Mitochondrial Heterogeneity as It Exists under Physiological States
3.1. Membrane Potential Heterogeneity Reveals Mitochondrial Subpopulations
3.2. Mitochondrial Morphology Influences Metabolic Signaling and Nutrient Sensitivity
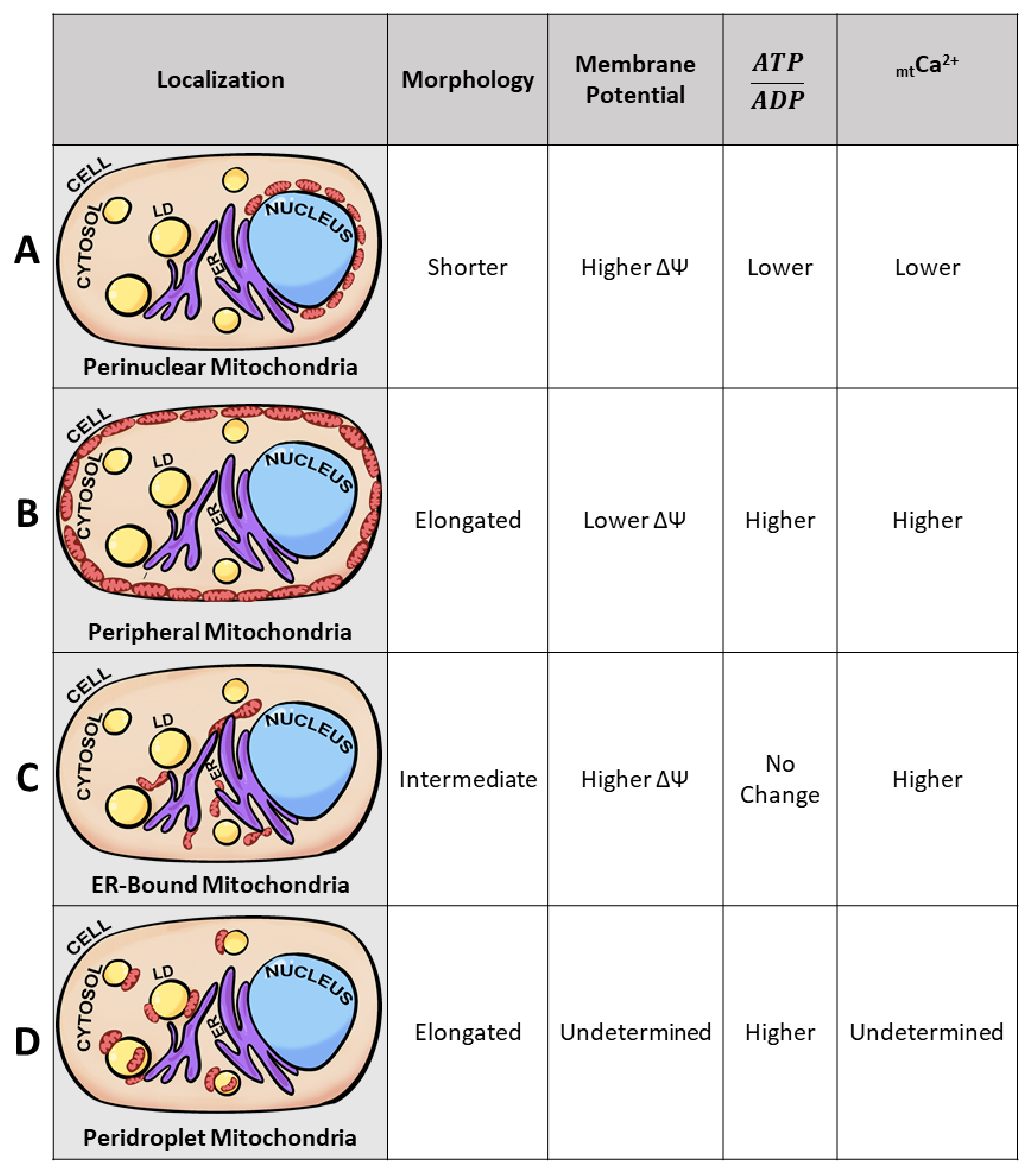
3.3. Mitochondrial Calcium as an Intracellular Heterogeneity Amplifier in Morphology and Motility
3.4. Mitochondrial Subpopulations Influence Cellular Function
3.5. Mitochondrial Heteroplasmy Potentially Promote Formation of Mitochondrial Subpopulations
4. Discussion
4.1. The Advantages of Mitochondrial Heterogeneity
4.2. Challenges and Considerations to Be Made When Studying Intracellular Heterogeneity
4.3. Remaining Questions in the Field
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aryaman, J.; Johnston, I.G.; Jones, N.S. Mitochondrial heterogeneity. Front. Genet. 2019, 9, 718. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Troppmair, J.; Sucher, R.; Hermann, M.; Saks, V.; Margreiter, R. Mitochondrial subpopulations and heterogeneity revealed by confocal imaging: Possible physiological role? Biochim. Biophys. Acta-Bioenerg. 2006, 1757, 686–691. [Google Scholar] [CrossRef]
- Wikstrom, J.D.; Twig, G.; Shirihai, O.S. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int. J. Biochem. Cell Biol. 2009, 41, 1914–1927. [Google Scholar] [CrossRef]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef]
- Wikstrom, J.D.; Katzman, S.M.; Mohamed, H.; Twig, G.; Graf, S.A.; Heart, E.; Molina, A.J.A.; Corkey, B.E.; de Vargas, L.M.; Danial, N.N.; et al. β-Cell Mitochondria Exhibit Membrane Potential Heterogeneity That Can Be Altered by Stimulatory or Toxic Fuel Levels. Diabetes 2007, 56, 2569–2578. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef]
- Duchen, M.R.; Surin, A.; Jacobson, J. Imaging mitochondrial function in intact cells. Methods Enzymol. 2003, 361, 353–389. [Google Scholar] [CrossRef]
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019, 38, e101056. [Google Scholar] [CrossRef] [PubMed]
- Zorzano, A.; Liesa, M.; Sebastián, D.; Segalés, J.; Palacín, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 1092–1097. [Google Scholar] [CrossRef]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical differences between subsarcolemmal and interfibrillar mitochondria from rat cardiac muscle: Effects of procedural manipulations. Arch. Biochem. Biophys. 2004, 236, 691–702. [Google Scholar] [CrossRef]
- Herms, A.; Bosch, M.; Ariotti, N.; Reddy, B.J.N.; Fajardo, A.; Fernández-Vidal, A.; Alvarez-Guaita, A.; Fernández-Rojo, M.A.; Rentero, C.; Tebar, F.; et al. Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Curr. Biol. 2013, 23, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Stotland, A. MitoTimer: A novel protein for monitoring mitochondrial turnover in the heart. J. Mol. Med. 2015, 93, 271–278. [Google Scholar] [CrossRef]
- Georgiadou, E.; Muralidharan, C.; Martinez, M.; Chabosseau, P.; Tomas, A.; Wern, F.Y.S.; Stylianides, T.; Rothery, S.M.; Di Gregorio, A.; Leclerc, I.; et al. Pancreatic beta cell selective deletion of mitofusins 1 and 2 (Mfn1 and Mfn2) disrupts mitochondrial architecture and abrogates glucose-stimulated insulin secretion in vivo. bioRxiv 2020. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Ward, M.W. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 2000, 23, 166–174. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Beraud, N.; Pelloux, S.; Usson, Y.; Kuznetsov, A.V.; Ronot, X.; Tourneur, Y.; Saks, V. Mitochondrial dynamics in heart cells: Very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J. Bioenerg. Biomembr. 2009, 41, 195–214. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Schneeberger, S.; Renz, O.; Meusburger, H.; Saks, V.; Usson, Y.; Margreiter, R. Functional heterogeneity of mitochondria after cardiac cold ischemia and reperfusion revealed by confocal imaging. Transplantation 2004, 77, 754–756. [Google Scholar] [CrossRef]
- Molina, A.J.A.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects β-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef]
- Wiederkehr, A.; Wollheim, C.B. Mitochondrial signals drive insulin secretion in the pancreatic β-cell. Mol. Cell. Endocrinol. 2012, 353, 128–137. [Google Scholar] [CrossRef]
- Heart, E.; Corkey, R.F.; Wikstrom, J.D.; Shirihai, O.S.; Corkey, B.E. Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells. Am. J. Physiol. Metab. 2006, 290, E143–E148. [Google Scholar] [CrossRef]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyon, J.Y.; Min, J.Y.; Huh, Y.H.; Kim, H.J.; Lee, H.; Yun, S.H.; Choi, C.W.; Jeong Ha, S.; Park, J.; et al. Mitochondrial carnitine palmitoyltransferase 2 is involved in Nε-(carboxymethyl)-lysine-mediated diabetic nephropathy. Pharmacol. Res. 2020, 152, 104600. [Google Scholar] [CrossRef]
- Kodiha, M.; Flamant, E.; Wang, Y.M.; Stochaj, U. Defining the short-term effects of pharmacological 50-AMP activated kinase modulators on mitochondrial polarization, morphology and heterogeneity. Peer J. 2018, 2018. [Google Scholar] [CrossRef]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.; Waggoner, J.; Cui, J.; et al. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; Bliek, A.M. van der The Novel Tail-anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta-Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Iqbal, S.; Hood, D.A. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol.-Cell Physiol. 2014, 306, C1176–C1183. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Stiles, L.; Shirihai, O.S. Mitochondrial dynamics and morphology in beta-cells. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 725–738. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Palacín, M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int. J. Biochem. Cell Biol. 2009, 41, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Nakatsuka, A. Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med. Okayama 2016, 70, 151–158. [Google Scholar]
- Donath, M.Y.; Halban, P.A. Decreased beta-cell mass in diabetes: Significance, mechanisms and therapeutic implications. Diabetologia 2004, 47, 581–589. [Google Scholar] [CrossRef]
- Nasteska, D.; Hodson, D.J. The role of beta cell heterogeneity in islet function and insulin release. J. Mol. Endocrinol. 2018, 61, R43–R60. [Google Scholar] [CrossRef]
- Men, X.; Wang, H.; Li, M.; Cai, H.; Xu, S.; Zhang, W.; Xu, Y.; Ye, L.; Yang, W.; Wollheim, C.B.; et al. Dynamin-related protein 1 mediates high glucose induced pancreatic beta cell apoptosis. Int. J. Biochem. Cell Biol. 2009, 41, 879–890. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2014, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G.; Bakajsova, D.; Samarel, A.M. Protein kinase C-εon induces mitochondrial dysfunction and fragmentation in renal proximal tubules. Am. J. Physiol.-Ren. Physiol. 2011, 301, F197–F208. [Google Scholar] [CrossRef]
- Jang, H.-S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Schuh, C.-D. Mitochondria as therapeutic targets in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2016, 25, 355–362. [Google Scholar] [CrossRef]
- Miguel, V.; Tituaña, J.; Herrero, J.I.; Herrero, L.; Serra, D.; Cuevas, P.; Barbas, C.; Puyol, D.R.; Márquez-Exposito, L.; Ruiz-Ortega, M.; et al. Renal tubule Cpt1a overexpression mitigates kidney fibrosis by restoring mitochondrial homeostasis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; Tengholm, A. Metabolic regulation of calcium signaling in beta cells. Semin. Cell Dev. Biol. 2020, 103, 20–30. [Google Scholar] [CrossRef]
- Gilon, P.; Chae, H.-Y.; Rutter, G.; Ravier, M.A. Calcium signaling in pancreatic β-cells in health and in Type 2 diabetes. Cell Calcium 2014, 56, 340–361. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Assali, E.A.; Jones, A.E.; Veliova, M.; Acín-Pérez, R.; Taha, M.; Miller, N.; Shum, N.; Oliveira, M.F.; Las, G.; Liesa, M.; et al. NCLX prevents cell death during adrenergic activation of the brown adipose tissue. Nat. Commun. 2004, 11, 3347. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Bailbé, D.; Gylfe, E.; Portha, B. Defective glucose-dependent cytosolic Ca2+ handling in islets of GK and nSTZ rat models of type 2 diabetes. J. Endocrinol. 2001, 169, 169–176. [Google Scholar] [CrossRef][Green Version]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Grodsky, G.M.; Bennett, L.L. Cation Requirements for Insulin Secretion in the Isolated Perfused Pancreas. Diabetes 1966, 15, 910. [Google Scholar] [CrossRef] [PubMed]
- Jahanshahi, P.; Wu, R.; Carter, J.D.; Nunemaker, C. Evidence of Diminished Glucose Stimulation and Endoplasmic Reticulum Function in Nonoscillatory Pancreatic Islets. Endocrinology 2009, 150, 607–615. [Google Scholar] [CrossRef][Green Version]
- Bitar, M.S.; Al-Saleh, E.; Al-Mulla, F. Oxidative stress—Mediated alterations in glucose dynamics in a genetic animal model of type II diabetes. Life Sci. 2005, 77, 2552–2573. [Google Scholar] [CrossRef]
- Rose, T.; Efendic, S.; Rupnik, M. Ca2+–Secretion Coupling Is Impaired in Diabetic Goto Kakizaki rats. J. Gen. Physiol. 2007, 129, 493–508. [Google Scholar] [CrossRef]
- Zaitsev, S.; Efanova, I.; Östenson, C.-G.; Efendić, S.; Berggren, P.-O. Delayed Ca2+Response to Glucose in Diabetic GK Rat. Biochem. Biophys. Res. Commun. 1997, 239, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Ishida, H.; Tsuura, Y.; Tsuji, K.; Nishimura, M.; Horie, M.; Taminato, T.; Ikehara, S.; Odaka, H.; Ikeda, I.; et al. Alterations in basal and glucose-stimulated voltage-dependent Ca2+ channel activities in pancreatic beta cells of non-insulin-dependent diabetes mellitus GK rats. J. Clin. Investig. 1996, 97, 2417–2425. [Google Scholar] [CrossRef] [PubMed]
- Portha, B.; Lacraz, G.; Kergoat, M.; Homo-Delarche, F.; Giroix, M.H.; Bailbé, D.; Gangnerau, M.N.; Dolz, M.; Tourrel-Cuzin, C.; Movassat, J. The GK rat beta-cell: A prototype for the diseased human beta-cell in type 2 diabetes? Mol. Cell. Endocrinol. 2009, 297, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Warnotte, C.; Gilon, P.; Nenquin, M.; Henquin, J.-C. Mechanisms of the Stimulation of Insulin Release by Saturated Fatty Acids: A Study of Palmitate Effects in Mouse β-cells. Diabetes 1994, 43, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, C.; Göpel, S.O.; Barg, S.; Galvanovskis, J.; Ma, X.; Salehi, A.; Rorsman, P.; Eliasson, L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Arch Eur. J. Physiol. 2002, 444, 43–51. [Google Scholar] [CrossRef]
- Nolan, C.J.; Madiraju, M.S.R.; Delghingaro-Augusto, V.; Peyot, M.-L.; Prentki, M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 2006, 55 (Suppl. 2), S16–S23. [Google Scholar] [CrossRef]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. et Biophys. Acta-Mol. Cell Biol. Lipids 2010, 1801, 289–298. [Google Scholar] [CrossRef]
- Tarasov, A.; Semplici, F.; Ravier, M.; Bellomo, E.; Pullen, T.; Gilon, P.; Sekler, I.; Rizzuto, R.; Rutter, G.A. The Mitochondrial Ca2+ Uniporter MCU Is Essential for Glucose-Induced ATP Increases in Pancreatic β-Cells. PLoS ONE 2012, 7, e39722. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman-Weinberg, D.; Kenyon, N.S.; Ricordi, C.; Berggren, P.-O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef]
- Pizzo, P.; Drago, I.; Filadi, R.; Pozzan, T. Mitochondrial Ca2+ homeostasis: Mechanism, role, and tissue specificities. Pflügers Arch.-Eur. J. Physiol. 2012, 464, 3–17. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. An intimate liaison: Spatial organization of the endoplasmic reticulum–mitochondria relationship. EMBO J. 2010, 29, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Hoppa, M.B.; Collins, S.; Ramracheya, R.; Hodson, L.; Amisten, S.; Zhang, Q.; Johnson, P.; Ashcroft, F.M.; Rorsman, P. Chronic Palmitate Exposure Inhibits Insulin Secretion by Dissociation of Ca2+ Channels from Secretory Granules. Cell Metab. 2009, 10, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.C.; Hoppa, M.B.; Walker, J.N.; Amisten, S.; Abdulkader, F.; Bengtsson, M.; Fearnside, J.; Ramracheya, R.; Toye, A.A.; Zhang, Q.; et al. Progression of Diet-Induced Diabetes in C57BL6J Mice Involves Functional Dissociation of Ca2+ Channels from Secretory Vesicles. Diabetes 2010, 59, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Mahdaviani, K.; Benador, I.Y.; Su, S.; Gharakhanian, R.; Stiles, L.; Trudeau, K.M.; Cardamone, M.; Enríquez-Zarralanga, V.; Ritou, E.; Aprahamian, T.; et al. Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenesis. EMBO Rep. 2017, 18, 1123–1138. [Google Scholar] [CrossRef]
- Leung, Y.M.; Ahmed, I.; Sheu, L.; Tsushima, R.G.; Diamant, N.E.; Hara, M.; Gaisano, H.Y. Electrophysiological Characterization of Pancreatic Islet Cells in the Mouse Insulin Promoter-Green Fluorescent Protein Mouse. Endocrinology 2005, 146, 4766–4775. [Google Scholar] [CrossRef]
- Park, M.K.; Ashby, M.; Erdemli, G.; Petersen, O.H.; Tepikin, A. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001, 20, 1863–1874. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of Mitochondria and Mitochondrial Function within Cells as Another Level of Mitochondrial Complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial oxidative function in NAFLD: Friend or foe? Mol. Metab. 2020, 50, 101134. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.; Benador, I.Y.; Brownstein, A.J.; Vergnes, L.; Veliova, M.; Shum, M.; Acín-Pérez, R.; Reue, K.; Shirihai, O.S.; Liesa, M. Isolation and functional analysis of peridroplet mitochondria from murine brown adipose tissue. STAR Protoc. 2021, 2, 100243. [Google Scholar] [CrossRef]
- Al-Mehdi, A.-B.; Pastukh, V.V.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear Mitochondrial Clustering Creates an Oxidant-Rich Nuclear Domain Required for Hypoxia-Induced Transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta-Bioenerg. 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B. Pathophysiological and protective roles of mitochondrial ion channels. J. Physiol. 2000, 529, 23–36. [Google Scholar] [CrossRef]
- Lonergan, T.; Bavister, B.; Brenner, C. Mitochondria in stem cells. Mitochondrion 2007, 7, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Freyre, C.A.; Rauher, P.C.; Ejsing, C.S.; Klemm, R.W. Faculty Opinions recommendation of MIGA2 Links Mitochondria, the ER, and Lipid Droplets and Promotes De Novo Lipogenesis in Adipocytes. Mol. Cell 2020, 76, 811–825. [Google Scholar] [CrossRef]
- Dzeja, P.P.; Bortolon, R.; Perez-Terzic, C.; Holmuhamedov, E.; Terzic, A. Energetic communication between mitochondria and nucleus directed by catalyzed phosphotransfer. Proc. Natl. Acad. Sci. USA 2002, 99, 10156–10161. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim Biophys Acta Gen. Subj. 2006, 1757, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.I.E.; Giovannucci, D.R.; Blinder, G.; Shuttleworth, T.J.; Yule, D.I. Modulation of [Ca2+] Signaling Dynamics and Metabolism by Perinuclear Mitochondria in Mouse Parotid Acinar Cells. J. Biol. Chem. 2004, 279, 12909–12917. [Google Scholar] [CrossRef]
- Yu, T.; Sheu, S.-S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef]
- Curthoys, N.P.; Moe, O.W. Proximal tubule function and response to acidosis. Clin. J. Am. Soc. Nephrol. 2014, 9, 1627–1638. [Google Scholar] [CrossRef]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Wieckowski, M.; Youle, R.J.; Rizzuto, R. Drp-1-Dependent Division of the Mitochondrial Network Blocks Intraorganellar Ca2+ Waves and Protects against Ca2+-Mediated Apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Frieden, M.; James, D.; Castelbou, C.; Danckaert, A.; Martinou, J.-C.; Demaurex, N. Ca2+ Homeostasis during Mitochondrial Fragmentation and Perinuclear Clustering Induced by hFis1. J. Biol. Chem. 2004, 279, 22704–22714. [Google Scholar] [CrossRef] [PubMed]
- Porat-Shliom, N.; Harding, O.J.; Malec, L.; Narayan, K.; Weigert, R. Mitochondrial Populations Exhibit Differential Dynamic Responses to Increased Energy Demand during Exocytosis In Vivo. iScience 2019, 11, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Noske, A.B.; Costin, A.; Morgan, G.P.; Marsh, B.J. Expedited approaches to whole cell electron tomography and organelle mark-up in situ in high-pressure frozen pancreatic islets. J. Struct. Biol. 2008, 161, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Renström, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef]
- Dean, P.M. Ultrastructural morphometry of the pancreatic β-cell. Diabetologia 1973, 9, 115–119. [Google Scholar] [CrossRef][Green Version]
- Stallons, L.J.; Funk, J.A.; Schnellmann, R.G. Mitochondrial Homeostasis in Acute Organ Failure. Curr. Pathobiol. Rep. 2013, 1, 169–177. [Google Scholar] [CrossRef]
- Hüser, J.; Blatter, L.A.; Sheu, S.S. Mitochondrial calcium in heart cells: Beat-to-beat oscillations or slow integration of cytosolic transients? J. Bioenerg. Biomembr. 2000, 32, 27–33. [Google Scholar] [CrossRef]
- Georgiadou, E.; Rutter, G.A. Control by Ca2+ of mitochondrial structure and function in pancreatic β-cells. Cell Calcium 2020, 91, 102282. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pi, H.; Chen, Y.; Zhang, N.; Guo, P.; Lu, Y.; He, M.; Xie, J.; Zhong, M.; Zhang, Y.; et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013, 4, e540. [Google Scholar] [CrossRef]
- Pennanen, C.; Parra, V.; López-Crisosto, C.; Morales, P.E.; del Campo, A.; Gutierrez, T.; Rivera-Mejías, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014, 127, 2659–2671. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Kremneva, E.; Kislin, M.; Kang, X.; Khiroug, L. Motility of astrocytic mitochondria is arrested by Ca2+-dependent interaction between mitochondria and actin filaments. Cell Calcium 2013, 53, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Saotome, M.; Safiulina, D.; Szabadkai, G.; Das, S.; Fransson, A.; Aspenstrom, P.; Rizzuto, R.; Hajnóczky, G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar] [CrossRef]
- Woods, D.C. Mitochondrial Heterogeneity: Evaluating Mitochondrial Subpopulation Dynamics in Stem Cells. Stem Cells Int. 2017, 2017, 1–7. [Google Scholar] [CrossRef]
- Bianchi, K.; Rimessi, A.; Prandini, A.; Szabadkai, G.; Rizzuto, R. Calcium and mitochondria: Mechanisms and functions of a troubled relationship. Biochim. Biophys. Acta-Mol. Cell Res. 2004, 1742, 119–131. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-Mitochondria Tethering Complex Revealed by a Synthetic Biology Screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated Analysis of Protein Composition, Tissue Diversity, and Gene Regulation in Mouse Mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Canto, C. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef]
- Taylor, S.W.; Fahy, E.; Zhang, B.; Glenn, G.M.; Warnock, D.E.; Wiley, S.; Murphy, A.N.; Gaucher, S.P.; Capaldi, R.A.; Gibson, B.W.; et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003, 21, 281–286. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel Excess and β-Cell Dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Frigerio, F.; Maechler, P. The sensitivity of pancreatic β-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem. Soc. Trans. 2008, 36, 930–934. [Google Scholar] [CrossRef]
- Grishko, V.; Rachek, L.; Musiyenko, S.; LeDoux, S.P.; Wilson, G.L. Involvement of mtDNA damage in free fatty acid-induced apoptosis. Free. Radic. Biol. Med. 2005, 38, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Jones, N. Discovering Cellular Mitochondrial Heteroplasmy Heterogeneity with Single Cell RNA and ATAC Sequencing. Biology 2021, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Narendra, D.P.; Tanaka, A.; Manfredi, G.; Youle, R.J. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11835–11840. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42. [Google Scholar] [CrossRef]
- Song, S.; Park, J.; Jang, S.; Hwang, E. Nicotinamide Treatment Facilitates Mitochondrial Fission through Drp1 Activation Mediated by SIRT1-Induced Changes in Cellular Levels of cAMP and Ca2+. Cells 2021, 10, 612. [Google Scholar] [CrossRef]
- Luan, P.; D’Amico, D.; Andreux, P.A.; Laurila, P.-P.; Wohlwend, M.; Li, H.; de Lima, T.I.; Place, N.; Rinsch, C.; Zanou, N.; et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci. Transl. Med. 2021, 13, eabb0319. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Keenan, S.; Meex, R.C.; Lo, J.C.Y.; Ryan, A.; Nie, S.; Montgomery, M.; Watt, M.J. Perilipin 5 Deletion in Hepatocytes Remodels Lipid Metabolism and Causes Hepatic Insulin Resistance in Mice. Diabetes 2019, 68, 543–555. [Google Scholar] [CrossRef]
- Trevino, M.B.; Mazur-Hart, D.; Machida, Y.; King, T.; Nadler, J.; Galkina, E.V.; Poddar, A.; Dutta, S.; Imai, Y. Liver Perilipin 5 Expression Worsens Hepatosteatosis But Not Insulin Resistance in High Fat-Fed Mice. Mol. Endocrinol. 2015, 29, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sreenivasan, U.; Hu, H.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res 2011, 52, 2159–2168. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2014, 61, 870–882. [Google Scholar] [CrossRef]
- Tan, J.; Jin, Y.; Wang, Q.; Huang, J.; Wu, X.; Ren, Z. Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells. Cells 2019, 8, 1241. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, J.; Osto, C.; Villalobos, F.; Shirihai, O.S. Mitochondrial Heterogeneity in Metabolic Diseases. Biology 2021, 10, 927. https://doi.org/10.3390/biology10090927
Ngo J, Osto C, Villalobos F, Shirihai OS. Mitochondrial Heterogeneity in Metabolic Diseases. Biology. 2021; 10(9):927. https://doi.org/10.3390/biology10090927
Chicago/Turabian StyleNgo, Jennifer, Corey Osto, Frankie Villalobos, and Orian S. Shirihai. 2021. "Mitochondrial Heterogeneity in Metabolic Diseases" Biology 10, no. 9: 927. https://doi.org/10.3390/biology10090927
APA StyleNgo, J., Osto, C., Villalobos, F., & Shirihai, O. S. (2021). Mitochondrial Heterogeneity in Metabolic Diseases. Biology, 10(9), 927. https://doi.org/10.3390/biology10090927