
Transposable Elements in the Genome of the Lichen-Forming Fungus Umbilicaria pustulata and Their Distribution in Different Climate Zones along Elevation

 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Genome of U. pustulata
2.2. Pool-Seq Sequencing of 15 U. pustulata Populations
2.3. De Novo TE Prediction: Building a U. pustulata TE Consensus Library
2.4. Evaluation of TE Copy Insertion Frequencies across the Different U. pustulata Populations
2.5. Identification of TE Loci Significantly Differentiated between U. pustulata Ecotypes
2.6. Functional Characterization
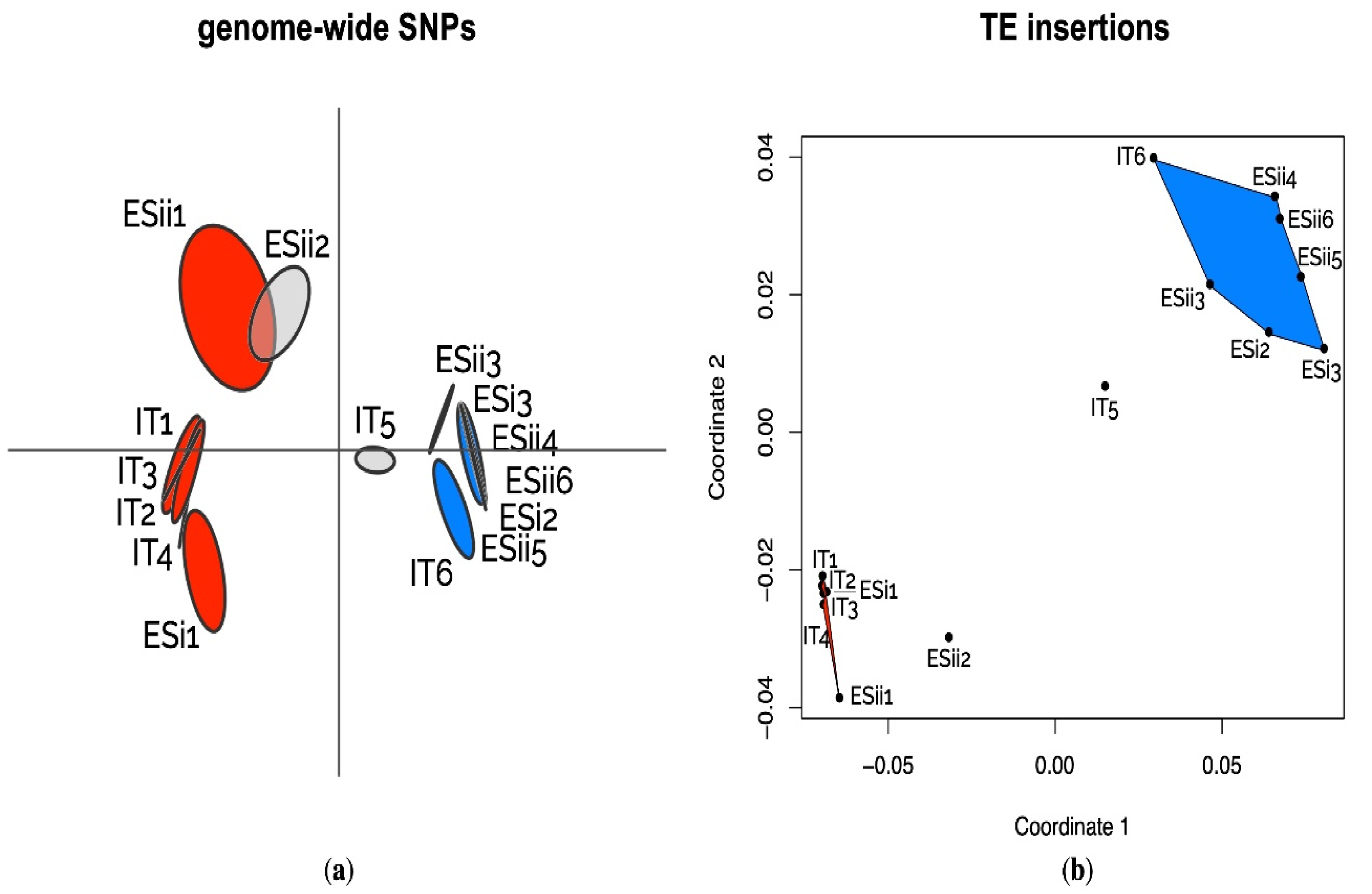
2.7. Population Structure Based on Genome-Wide SNPs
3. Results
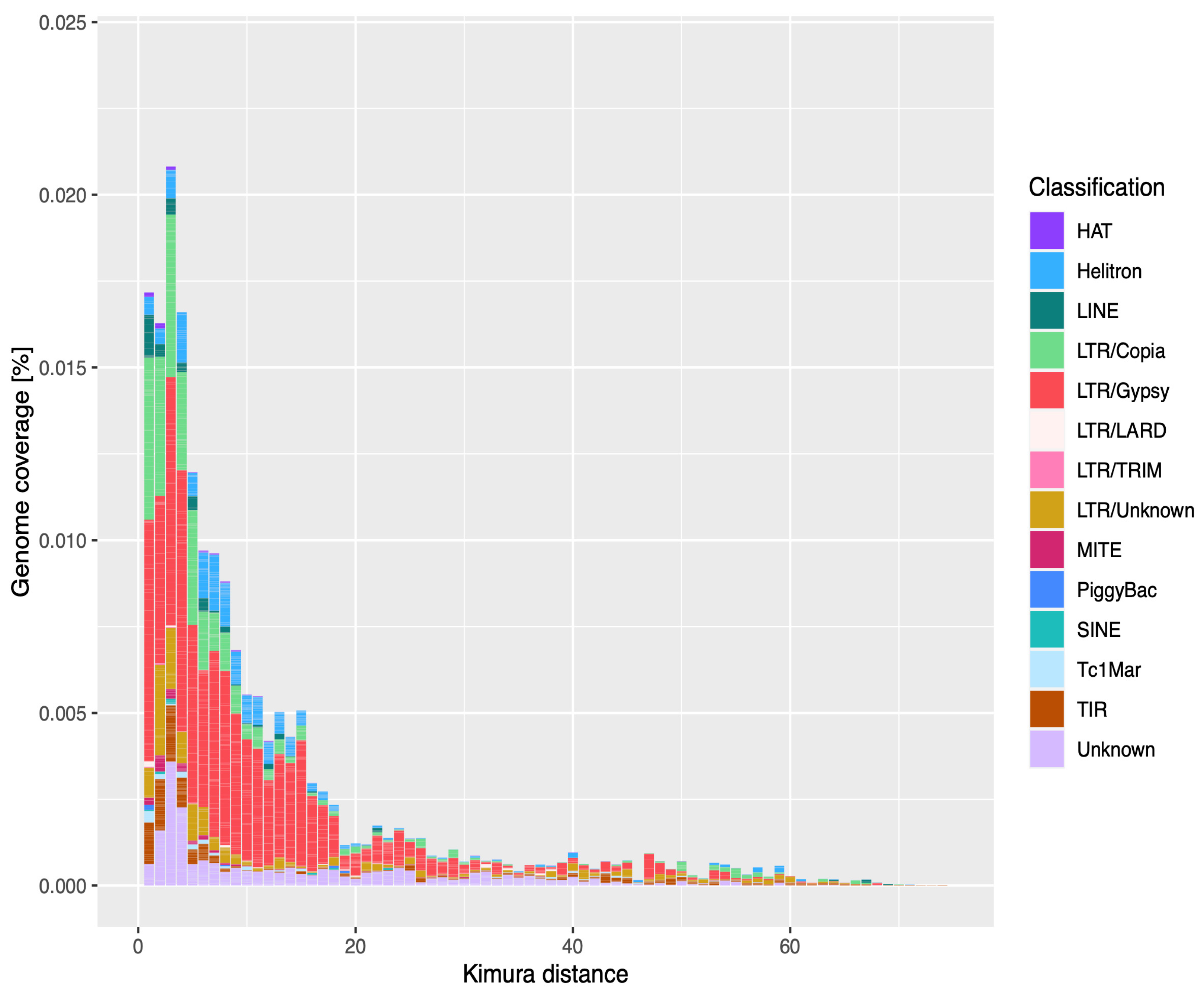
3.1. TE Landscape in U. pustulata
3.2. TE Variation across U. pustulata Populations
3.3. Variations of TE Frequencies between Ecotypes
3.4. Potential Functional Impact of TE Insertions
4. Discussion
4.1. The U. pustulata Mobilome
4.2. Ecotypic Differentiation Patterns of TE Insertions and Their Potential Functional Impact
4.3. Outlook and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sawyer, S.; Hartl, D. Distribution of transposable elements in prokaryotes. Theor. Popul. Biol. 1986, 30, 1–16. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr. Opin. Genet. Dev. 2005, 15, 621–627. [Google Scholar] [CrossRef]
- Staton, S.E.; Burke, J.M. Evolutionary transitions in the Asteraceae coincide with marked shifts in transposable element abundance. BMC Genom. 2015, 16, 623. [Google Scholar] [CrossRef] [Green Version]
- Daboussi, M.-J.; Capy, P. Transposable Elements in Filamentous Fungi. Annu. Rev. Microbiol. 2003, 57, 275–299. [Google Scholar] [CrossRef]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.-N. Comparative Analysis of Transposable Elements Highlights Mobilome Diversity and Evolution in Vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Armisén, D.; Gibbs, R.A.; Hering, L.; Khila, A.; Mayer, G.; Richards, S.; Niehuis, O.; Misof, B. Diversity and evolution of the transposable element repertoire in arthropods with particular reference to insects. BMC Ecol. Evol. 2019, 19, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef]
- Quadrana, L.; Etcheverry, M.; Gilly, A.; Caillieux, E.; Madoui, M.-A.; Guy, J.; Silveira, A.B.; Engelen, S.; Baillet, V.; Wincker, P.; et al. Transposition favors the generation of large effect mutations that may facilitate rapid adaption. Nat. Commun. 2019, 10, 3421. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 1999, 9, 657–663. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Boulesteix, M.; Weiss, M.; Biémont, C. Differences in Genome Size between Closely Related Species: The Drosophila melanogaster Species Subgroup. Mol. Biol. Evol. 2006, 23, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.J.; O’Neill, R.J. Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosom. Res. 2018, 26, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarevitch, I.; Waters, A.J.; West, P.T.; Stitzer, M.; Hirsch, C.N.; Ross-Ibarra, J.; Springer, N.M. Transposable Elements Contribute to Activation of Maize Genes in Response to Abiotic Stress. PLoS Genet. 2015, 11, e1004915. [Google Scholar] [CrossRef] [Green Version]
- Rey, O.; Danchin, E.; Mirouze, M.; Loot, C.; Blanchet, S. Adaptation to Global Change: A Transposable Element–Epigenetics Perspective. Trends Ecol. Evol. 2016, 31, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Pritham, E.J. DNA Transposons and the Evolution of Eukaryotic Genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [Green Version]
- Pritham, E.J. Transposable Elements and Factors Influencing their Success in Eukaryotes. J. Hered. 2009, 100, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Biémont, C.; Vieira, C. Genetics: Junk DNA as an evolutionary force. Nature 2006, 443, 521–524. [Google Scholar] [CrossRef]
- Schmidt, A.L.; Anderson, L.M. Repetitive DNA elements as mediators of genomic change in response to environmental cues. Biol. Rev. Camb. Philos. Soc. 2006, 81, 531–543. [Google Scholar] [CrossRef]
- Oliver, K.R.; Greene, W.K. Transposable elements: Powerful facilitators of evolution. BioEssays 2009, 31, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Hua-Van, A.; Le Rouzic, A.; Boutin, T.S.; Filée, J.; Capy, P. The struggle for life of the genome’s selfish architects. Biol. Direct 2011, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Casacuberta, E.; González, J. The impact of transposable elements in environmental adaptation. Mol. Ecol. 2013, 22, 1503–1517. [Google Scholar] [CrossRef]
- Hof, A.E.V.; Campagne, P.; Rigden, D.; Yung, C.J.; Lingley, J.; Quail, M.A.; Hall, N.; Darby, A.C.; Saccheri, I.J. The industrial melanism mutation in British peppered moths is a transposable element. Nature 2016, 534, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, L.; Schmitz, J. The impact of transposable elements in adaptive evolution. Mol. Ecol. 2019, 28, 1537–1549. [Google Scholar] [CrossRef]
- González, J.; Petrov, D.A. The adaptive role of transposable elements in the Drosophila genome. Gene 2009, 448, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, V.; Merenciano, M.; González, J. Revisiting the Relationship between Transposable Elements and the Eukaryotic Stress Response. Trends Genet. 2017, 33, 832–841. [Google Scholar] [CrossRef]
- Dubin, M.J.; Scheid, O.M.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef]
- Arkhipova, I.R. Neutral Theory, Transposable Elements, and Eukaryotic Genome Evolution. Mol. Biol. Evol. 2018, 35, 1332–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maumus, F.; Allen, A.E.; Mhiri, C.; Hu, H.; Jabbari, K.; Vardi, A.; Grandbastien, M.-A.; Bowler, C. Potential impact of stress activated retrotransposons on genome evolution in a marine diatom. BMC Genom. 2009, 10, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, K.; Cho, E.; Yang, G.; Campbell, M.A.; Yano, K.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Dramatic amplification of a rice transposable element during recent domestication. Proc. Natl. Acad. Sci. USA 2006, 103, 17620–17625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Levin, H.L. High-throughput sequencing of retrotransposon integration provides a saturated profile of target activity in Schizosaccharomyces pombe. Genome Res. 2009, 20, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef]
- Servant, G.; Pinson, B.; Tchalikian-Cosson, A.; Coulpier, F.; Lemoine, S.; Pennetier, C.; Bridier-Nahmias, A.; Todeschini, A.L.; Fayol, H.; Daignan-Fornier, B.; et al. Tye7 regulates yeast Ty1 retrotransposon sense and antisense transcription in response to adenylic nucleotides stress. Nucleic Acids Res. 2012, 40, 5271–5282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Xie, W.; Brady, T.L.; Gao, J.; Voytas, D.F. Phosphorylation Regulates Integration of the Yeast Ty5 Retrotransposon into Heterochromatin. Mol. Cell 2007, 27, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lockton, S.; Ross-Ibarra, J.; Gaut, B.S. Demography and weak selection drive patterns of transposable element diversity in natural populations of Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2008, 105, 13965–13970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.; Kural, D.; Strömberg, M.P.; Walker, J.A.; Konkel, M.K.; Stütz, A.M.; Urban, A.E.; Grubert, F.; Lam, H.Y.K.; Lee, W.-P.; et al. A Comprehensive Map of Mobile Element Insertion Polymorphisms in Humans. PLoS Genet. 2011, 7, e1002236. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-W.; Hou, X.-H.; Chen, J.-F.; Xu, Y.-C.; Wu, Q.; González, J.; Guo, Y.-L. Transposable Elements Contribute to the Adaptation of Arabidopsis thaliana. Genome Biol. Evol. 2018, 10, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Wos, G.; Choudhury, R.R.; Kolář, F.; Parisod, C. Transcriptional activity of transposable elements along an elevational gradient in Arabidopsis arenosa. Mob. DNA 2021, 12, 7. [Google Scholar] [CrossRef]
- Pietzenuk, B.; Markus, C.; Gaubert, H.; Bagwan, N.; Merotto, A.; Bucher, E.; Pecinka, A. Recurrent evolution of heat-responsiveness in Brassicaceae COPIA elements. Genome Biol. 2016, 17, 209. [Google Scholar] [CrossRef] [Green Version]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Major ecological adaptations and evolutionary transitions. Biol. Rev. 2019, 94, 1443–1476. [Google Scholar] [CrossRef] [Green Version]
- Abrego, N.; Crosier, B.; Somervuo, P.; Ivanova, N.; Abrahamyan, A.; Abdi, A.; Hämäläinen, K.; Junninen, K.; Maunula, M.; Purhonen, J.; et al. Fungal communities decline with urbanization—More in air than in soil. ISME J. 2020, 14, 2806–2815. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proc. Online 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorrain, C.; Feurtey, A.; Möller, M.; Haueisen, J.; Stukenbrock, E. Dynamics of transposable elements in recently diverged fungal pathogens: Lineage-specific transposable element content and efficiency of genome defenses. G3 Genes Genom. Genet. 2021, 11, jkab068. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, C.A.; Güldener, U.; Xu, J.-R.; Trail, F.; Turgeon, B.G.; Di Pietro, A.; Walton, J.D.; Ma, L.-J.; Baker, S.; Rep, M.; et al. The Fusarium graminearum Genome Reveals a Link between Localized Polymorphism and Pathogen Specialization. Science 2007, 317, 1400–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantzeskakis, L.; Kracher, B.; Kusch, S.; Yoshikawa-Maekawa, M.; Bauer, S.; Pedersen, C.; Spanu, P.D.; Maekawa, T.; Schulze-Lefert, P.; Panstruga, R. Signatures of host specialization and a recent transposable element burst in the dynamic one-speed genome of the fungal barley powdery mildew pathogen. BMC Genom. 2018, 19, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanera, R.; López-Varas, L.; Borgognone, A.; LaButti, K.; Lapidus, A.; Schmutz, J.; Grimwood, J.; Pérez, G.; Pisabarro, A.G.; Grigoriev, I.V.; et al. Transposable Elements versus the Fungal Genome: Impact on Whole-Genome Architecture and Transcriptional Profiles. PLoS Genet. 2016, 12, e1006108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oggenfuss, U.; Badet, T.; Wicker, T.; Hartmann, F.E.; Singh, N.K.; Abraham, L.; Karisto, P.; Vonlanthen, T.; Mundt, C.; McDonald, B.A.; et al. A population-level invasion by transposable elements triggers genome expansion in a fungal pathogen. eLife 2021, 10, e69249. [Google Scholar] [CrossRef]
- Grandaubert, J.; Lowe, R.G.; Soyer, J.L.; Schoch, C.L.; Van de Wouw, A.P.; Fudal, I.; Robbertse, B.; Lapalu, N.; Links, M.G.; Ollivier, B.; et al. Transposable element-assisted evolution and adaptation to host plant within the Leptosphaeria maculans-Leptosphaeria biglobosa species complex of fungal pathogens. BMC Genom. 2014, 15, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouché, S.; Plissonneau, C.; Croll, D. The birth and death of effectors in rapidly evolving filamentous pathogen genomes. Curr. Opin. Microbiol. 2018, 46, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Fokkens, L.; Shahi, S.; Connolly, L.R.; Stam, R.; Schmidt, S.M.; Smith, K.M.; Freitag, M.; Rep, M. The multi-speed genome of Fusarium oxysporum reveals association of histone modifications with sequence divergence and footprints of past horizontal chromosome transfer events. BioRxiv 2018, 465070. [Google Scholar] [CrossRef] [Green Version]
- Lutzoni, F.; Pagel, M.; Reeb, V. Major fungal lineages are derived from lichen symbiotic ancestors. Nature 2001, 411, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.A.; Peršoh, D.; Rambold, G. Umbilicariaceae (lichenized Ascomycota)—Trait evolution and a new generic concept. Taxon 2017, 66, 1282–1303. [Google Scholar] [CrossRef]
- Dal Grande, F.; Sharma, R.; Meiser, A.; Rolshausen, G.; Büdel, B.; Mishra, B.; Thines, M.; Otte, J.; Pfenninger, M.; Schmitt, I. Adaptive differentiation coincides with local bioclimatic conditions along an elevational cline in populations of a lichen-forming fungus. BMC Evol. Biol. 2017, 17, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Grande, F.; Rolshausen, G.; Divakar, P.K.; Crespo, A.; Otte, J.; Schleuning, M.; Schmitt, I. Environment and host identity structure communities of green algal symbionts in lichens. New Phytol. 2018, 217, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greshake-Tzovaras, B.; Segers, F.H.I.D.; Bicker, A.; Dal Grande, F.; Otte, J.; Anvar, S.Y.; Hankeln, T.; Schmitt, I.; Ebersberger, I. What Is in Umbilicaria pustulata? A Metagenomic Approach to Reconstruct the Holo-Genome of a Lichen. Genome Biol. Evol. 2020, 12, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofler, R.; Orozco-Terwengel, P.; De Maio, N.; Pandey, R.V.; Nolte, V.; Futschik, A.; Kosiol, C.; Schlötterer, C. PoPoolation: A Toolbox for Population Genetic Analysis of Next Generation Sequencing Data from Pooled Individuals. PLoS ONE 2011, 6, e15925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesneville, H.; Bergman, C.; Andrieu, O.; Autard, D.; Nouaud, D.; Ashburner, M.; Anxolabehere, D. Combined Evidence Annotation of Transposable Elements in Genome Sequences. PLoS Comput. Biol. 2005, 1, e22. [Google Scholar] [CrossRef] [PubMed]
- Flutre, T.; Duprat, E.; Feuillet, C.; Quesneville, H. Considering Transposable Element Diversification in De Novo Annotation Approaches. PLoS ONE 2011, 6, e16526. [Google Scholar] [CrossRef]
- Hoede, C.; Arnoux, S.; Moissette, M.; Chaumier, T.; Inizan, O.; Jamilloux, V.; Quesneville, H. PASTEC: An Automatic Transposable Element Classification Tool. PLoS ONE 2014, 9, e91929. [Google Scholar] [CrossRef] [Green Version]
- Jamilloux, V.; Daron, J.; Choulet, F.; Quesneville, H. De Novo Annotation of Transposable Elements: Tackling the Fat Genome Issue. Proc. IEEE 2017, 105, 474–481. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Gómez-Sánchez, D.; Schlötterer, C. PoPoolationTE2: Comparative Population Genomics of Transposable Elements Using Pool-Seq. Mol. Biol. Evol. 2016, 33, 2759–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.; Anders, S.; Huber, W. Differential analysis of count data—The DESeq2 package. Genome Biol. 2014, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Abdi, H.; O’Toole, A.J.; Valentin, D.; Edelman, B. DISTATIS: The analysis of multiple distance matrices. In Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’05)—Workshops IEEE, San Diego, CA, USA, 21–23 September 2005; p. 42. [Google Scholar] [CrossRef]
- Kofler, R.; Pandey, R.V.; Schlötterer, C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 2011, 27, 3435–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuomo, C.A.; Birren, B.W. The Fungal Genome Initiative and Lessons Learned from Genome Sequencing. Methods Enzymol. 2010, 470, 833–855. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Liu, B.; Zhang, X.-Y.; Zhou, Q.-M.; Zhang, T.; Li, H.; Yu, Y.-F.; Zhang, X.-L.; Hao, X.-Y.; Wang, M.; et al. Genome characteristics reveal the impact of lichenization on lichen-forming fungus Endocarpon pusillum Hedwig (Verrucariales, Ascomycota). BMC Genom. 2014, 15, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerat, E.; Goubert, C.; Guirao-Rico, S.; Merenciano, M.; Dufour, A.; Vieira, C.; González, J. Population-specific dynamics and selection patterns of transposable element insertions in European natural populations. Mol. Ecol. 2019, 28, 1506–1522. [Google Scholar] [CrossRef]
- Kozlowski, D.K.L.; Hassanaly-Goulamhoussen, R.; Da Rocha, M.; Koutsovoulos, G.D.; Bailly-Bechet, M.; Danchin, E.G.J. Movements of transposable elements contribute to the genomic plasticity and species diversification in an asexually reproducing nematode pest. Evol. Appl. 2021, 14, 1844–1866. [Google Scholar] [CrossRef] [PubMed]
- Muszewska, A.; Steczkiewicz, K.; Stepniewska-Dziubinska, M.; Ginalski, K. Transposable elements contribute to fungal genes and impact fungal lifestyle. Sci. Rep. 2019, 9, 4307. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable Elements: An Abundant and Natural Source of Regulatory Sequences for Host Genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef]
- Muszewska, A.; Steczkiewicz, K.; Stepniewska-Dziubinska, M.; Ginalski, K. Cut-and-paste transposons in fungi with diverse lifestyles. Genome Biol. Evol. 2017, 9, 3463–3477. [Google Scholar] [CrossRef] [Green Version]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Kawakami, T.; Dhakal, P.; Katterhenry, A.N.; Heatherington, C.A.; Ungerer, M.C. Transposable Element Proliferation and Genome Expansion Are Rare in Contemporary Sunflower Hybrid Populations Despite Widespread Transcriptional Activity of LTR Retrotransposons. Genome Biol. Evol. 2011, 3, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Waminal, N.; Choi, H.-I.; Perumal, S.; Lee, S.-C.; Nguyen, V.B.; Jang, W.; Kim, N.-H.; Gao, L.-Z.; Yang, T.-J. Rapid amplification of four retrotransposon families promoted speciation and genome size expansion in the genus Panax. Sci. Rep. 2017, 7, 9045. [Google Scholar] [CrossRef] [Green Version]
- Marcon, H.S.; Domingues, D.S.; Silva, J.C.; Borges, R.J.; Matioli, F.F.; Fontes, M.R.D.M.; Marino, C.L. Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol. 2015, 15, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essen, L.-O.; Vogt, M.S.; Mösch, H.-U. Diversity of GPI-anchored fungal adhesins. Biol. Chem. 2020, 401, 1389–1405. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Schmelzle, T.; Hall, M.N. The RHO1-GAPs SAC7, BEM2 and BAG7 control distinct RHO1 functions in Saccharomyces cerevisiae. Mol. Microbiol. 2002, 45, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Downes, D.; Davis, M.A.; Kreutzberger, S.D.; Taig, B.L.; Todd, R.B. Regulation of the NADP-glutamate dehydrogenase gene gdhA in Aspergillus nidulans by the Zn(II)2Cys6 transcription factor LeuB. Microbiology 2013, 159, 2467–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayerhofer, M.S.; Fraser, E.; Kernaghan, G. Acid protease production in fungal root endophytes. Mycologia 2015, 107, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.P.; Gillaspy, G.; Perera, I. Biosynthesis and possible functions of inositol pyrophosphates in plants. Front. Plant Sci. 2015, 6, 67. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Fabro, G.; Alvarez, M.E. Loss of compatibility might explain resistance of the Arabidopsis thaliana accession Te-0 to Golovinomyces cichoracearum. BMC Plant Biol. 2012, 12, 143. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Miguel-Rojas, C.; Lopez-Giraldez, F.; Yarden, O.; Trail, F.; Townsend, J.P. Metabolism and Development during Conidial Germination in Response to a Carbon-Nitrogen-Rich Synthetic or a Natural Source of Nutrition in Neurospora crassa. mBio 2019, 10, e00192-19. [Google Scholar] [CrossRef] [Green Version]
- Singh, G.; Calchera, A.; Schulz, M.; Drechsler, M.; Bode, H.B.; Schmitt, I.; Dal Grande, F. Climate-specific biosynthetic gene clusters in populations of a lichen-forming fungus. Environ. Microbiol. 2021, 23, 4260–4275. [Google Scholar] [CrossRef]
- Shaaban, M.; Palmer, J.M.; El-Naggar, W.A.; El-Sokkary, M.A.; Habib, E.-S.E.; Keller, N.P. Involvement of transposon-like elements in penicillin gene cluster regulation. Fungal Genet. Biol. 2010, 47, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Bast, J.; Jaron, K.S.; Schuseil, D.; Roze, D.; Schwander, T. Asexual reproduction reduces transposable element load in experimental yeast populations. eLife 2019, 8, e48548. [Google Scholar] [CrossRef]
- Bast, J.; Schaefer, I.; Schwander, T.; Maraun, M.; Scheu, S.; Kraaijeveld, K. No Accumulation of Transposable Elements in Asexual Arthropods. Mol. Biol. Evol. 2016, 33, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Szitenberg, A.; Cha, S.; Opperman, C.H.; Bird, D.M.; Blaxter, M.L.; Lunt, D.H. Genetic Drift, Not Life History or RNAi, Determine Long-Term Evolution of Transposable Elements. Genome Biol. Evol. 2016, 8, 2964–2978. [Google Scholar] [CrossRef] [Green Version]
- Poelt, J. Das Konzept der Artenpaare bei den Flechten. Vor aus dem Gesamtgebiet der Bot NF. Deutsche Botanische Gesellschaft 1970, 4, 187–198. [Google Scholar]
- Singh, G.; Dal Grande, F.; Cornejo, C.; Schmitt, I.; Scheidegger, C. Genetic Basis of Self-Incompatibility in the Lichen-Forming Fungus Lobaria pulmonaria and Skewed Frequency Distribution of Mating-Type Idiomorphs: Implications for Conservation. PLoS ONE 2012, 7, e51402. [Google Scholar] [CrossRef]
- Sancho, L.G.; Crespo, A. Lasallia hispanica and Related Species. Lichenologist 1989, 21, 45–58. [Google Scholar] [CrossRef]
- Dal Grande, F.; Meiser, A.; Greshake-Tzovaras, B.; Otte, J.; Ebersberger, I.; Schmitt, I. The draft genome of the lichen-forming fungus Lasallia hispanica (Frey) Sancho & A. Crespo. Lichenologist 2018, 50, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.; Skrede, I.; Wolfe, B.E.; LaButti, K.; Ohm, R.A.; Grigoriev, I.V.; Pringle, A. Transposable Element Dynamics among Asymbiotic and Ectomycorrhizal Amanita Fungi. Genome Biol. Evol. 2014, 6, 1564–1578. [Google Scholar] [CrossRef] [Green Version]
- Dutheil, J.Y.; Mannhaupt, G.; Schweizer, G.; Sieber, C.M.; Münsterkötter, M.; Güldener, U.; Schirawski, J.; Kahmann, R. A Tale of Genome Compartmentalization: The Evolution of Virulence Clusters in Smut Fungi. Genome Biol. Evol. 2016, 8, 681–704. [Google Scholar] [CrossRef] [Green Version]
- Shirke, M.D.; Mahesh, H.B.; Gowda, M. Genome-Wide Comparison of Magnaporthe Species Reveals a Host-Specific Pattern of Secretory Proteins and Transposable Elements. PLoS ONE 2016, 11, e0162458. [Google Scholar] [CrossRef]
- Chen, J.; Werth, S.; Sork, V.L. Comparison of phylogeographical structures of a lichen-forming fungus and its green algal photobiont in western North America. J. Biogeogr. 2016, 43, 932–943. [Google Scholar] [CrossRef]
- Worden, A.Z.; Lee, J.-H.; Mock, T.; Rouzé, P.; Simmons, M.P.; Aerts, A.L.; Allen, A.E.; Cuvelier, M.L.; Derelle, E.; Everett, M.V.; et al. Green Evolution and Dynamic Adaptations Revealed by Genomes of the Marine Picoeukaryotes Micromonas. Science 2009, 324, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Philippsen, G.S.; Avaca-Crusca, J.S.; Araujo, A.P.U.; DeMarco, R. Distribution patterns and impact of transposable elements in genes of green algae. Gene 2016, 594, 151–159. [Google Scholar] [CrossRef]
- Derelle, E.; Ferraz, C.; Rombauts, S.; Rouzé, P.; Worden, A.Z.; Robbens, S.; Partensky, F.; Degroeve, S.; Echeynié, S.; Cooke, R.; et al. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc. Natl. Acad. Sci. USA 2006, 103, 11647–11652. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Mendoza, F.; Domaschke, S.; García, M.A.; Jordan, P.; Martín, M.P.; Printzen, C. Population structure of mycobionts and photobionts of the widespread lichen Cetraria aculeata. Mol. Ecol. 2011, 20, 1208–1232. [Google Scholar] [CrossRef]
- Widmer, I.; Dal Grande, F.; Excoffier, L.; Holderegger, R.; Keller, C.; Mikryukov, V.S.; Scheidegger, C. European phylogeography of the epiphytic lichen fungus Lobaria pulmonaria and its green algal symbiont. Mol. Ecol. 2012, 21, 5827–5844. [Google Scholar] [CrossRef]
- Dal Grande, F.; Beck, A.; Cornejo, C.; Singh, G.; Cheenacharoen, S.; Nelsen, M.; Scheidegger, C. Molecular phylogeny and symbiotic selectivity of the green algal genus Dictyochloropsis s.l. (Trebouxiophyceae): A polyphyletic and widespread group forming photobiont-mediated guilds in the lichen family Lobariaceae. New Phytol. 2014, 202, 455–470. [Google Scholar] [CrossRef]
- Werth, S.; Sork, V.L. Ecological specialization in Trebouxia (Trebouxiophyceae) photobionts of Ramalina menziesii (Ramalinaceae) across six range-covering ecoregions of western North America. Am. J. Bot. 2014, 101, 1127–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolshausen, G.; Dal Grande, F.; Sadowska-Deś, A.D.; Otte, J.; Schmitt, I. Quantifying the climatic niche of symbiont partners in a lichen symbiosis indicates mutualist-mediated niche expansions. Ecography 2018, 41, 1380–1392. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Country | Population ID | Lat | Long | Elevation m a.s.l. | No. Paired-End Read # | Mean Read Length |
|---|---|---|---|---|---|---|
| Italy | IT1 | 40.7577 | 9.0794 | 176 | 29,162,770 | 99.3 |
| IT2 | 40.7778 | 9.0546 | 297 | 28,279,628 | 99.3 | |
| IT3 | 40.8503 | 9.1119 | 588 | 26,570,943 | 99.4 | |
| IT4 | 40.8568 | 9.1340 | 842 | 31,720,828 | 99.4 | |
| IT5 | 40.8573 | 9.1642 | 1125 | 31,755,901 | 99.4 | |
| IT6 | 40.8524 | 9.1732 | 1303 | 32,064,853 | 99.4 | |
| Spain 1 | ESii1 | 40.2028 | −5.2334 | 706 | 26,758,269 | 141.8 |
| ESii2 | 40.2069 | −5.2327 | 887 | 24,295,101 | 141.7 | |
| ESii3 | 40.2116 | −5.2337 | 1082 | 29,236,274 | 141.9 | |
| ESii4 | 40.2183 | −5.2335 | 1258 | 33,333,561 | 141.6 | |
| ESii5 | 40.2253 | −5.2375 | 1480 | 24,672,545 | 141.7 | |
| ESii6 | 40.2322 | −5.2389 | 1699 | 26,690,508 | 141.5 | |
| Spain 2 | ESi1 | 39.9946 | −4.8679 | 477 | 28,862,057 | 99.5 |
| ESi2 | 40.2899 | −4.9927 | 859 | 37,303,042 | 99.5 | |
| ESi3 | 40.3230 | −5.0173 | 1417 | 35,351,050 | 99.5 |
| A. Summary of Class I and II TE elements found in the U. pustulata genome. | |||||||||||
| Class | Total Length | No. Copies | No. Full Length Copies | Median Identity 1 | Median Length | ||||||
| Class II | 1,146,170 | 1863 | 156 | 91.4 | 657.9 | ||||||
| Class I | 5,118,614 | 2902 | 465 | 90.3 | 1162.5 | ||||||
| unknown | 731,643 | 1191 | 83 | 88.1 | 323.4 | ||||||
| B. Summary of TE elements subdivided into superfamilies for the U. pustulata genome. | |||||||||||
| Class | Order | Superfamily | No. Elements | Total Length | No. Copies | No. Full Length Copies | Median Identity 1 | Median Length | |||
| Class II | DHX | Helitron_01 | 7 | 553,513 | 680 | 23 | 88.7 | 498.6 | |||
| DTA | HAT | 1 | 24,206 | 80 | 4 | 89.98 | 186.5 | ||||
| DTB | PiggyBac | 1 | 12,236 | 10 | 4 | 95.3 | 1481.0 | ||||
| DTT | Tc1Mar | 4 | 104,574 | 139 | 28 | 89.6 | 1029.4 | ||||
| DTX | TIR | 18 | 380,415 | 824 | 86 | 92.0 | 648.2 | ||||
| DXX | MITE | 4 | 71,226 | 130 | 11 | 93.0 | 521.0 | ||||
| Class I | RII + RIX | LINE | 5 | 317,234 | 155 | 33 | 94.0 | 923.1 | |||
| RLC | Copia | 25 | 1,333,809 | 865 | 166 | 92.0 | 1350.2 | ||||
| RLG | Gypsy | 23 | 2,904,582 | 1296 | 215 | 89.8 | 1246.0 | ||||
| RLX | LTR | 15 | 538,504 | 550 | 46 | 86.2 | 942.6 | ||||
| RXX | LARD | 1 | 20,415 | 25 | 1 | 816.6 | 383.0 | ||||
| RXX | TRIM | 1 | 4070 | 11 | 4 | 96.8 | 126.0 | ||||
| No | Unknown | 14 | 731,643 | 1191 | 83 | 88.1 | 323.4 | ||||
| total | 119 | 6,996,427 | 5956 | 704 | 147.1 | 743.0 | |||||
| A. TE copy insertion in 15 populations of U. pustulata (min. physical coverage: 16×). | ||
| TE Family | Copy No. | % |
| Copia | 62 | 34.1 |
| TIR | 31 | 17.0 |
| Unknown | 23 | 12.6 |
| Helitron | 22 | 12.1 |
| Gypsy | 16 | 8.8 |
| LTR | 10 | 5.5 |
| MITE | 8 | 4.4 |
| LARD | 5 | 2.7 |
| TC1Mar | 2 | 1.1 |
| HAT | 1 | 0.5 |
| LINE | 1 | 0.5 |
| Piggybac | 1 | 0.5 |
| B. Polymorphic TE copy insertion in populations. | ||
| TE Family | Copy No. | % |
| Copia | 49 | 43.0 |
| TIR | 22 | 19.3 |
| Unknown | 13 | 11.4 |
| Helitron | 10 | 8.8 |
| Gypsy | 5 | 4.4 |
| LTR | 5 | 4.4 |
| MITE | 5 | 4.4 |
| LARD | 1 | 0.9 |
| TC1Mar | 2 | 1.8 |
| HAT | 1 | 0.9 |
| Piggybac | 1 | 0.9 |
| C. hdTEs between U. pustulata ecotypes. | ||
| TE Family | Copy No. | % |
| Copia | 16 | 57.1 |
| TIR | 4 | 14.3 |
| Helitron | 3 | 10.7 |
| Unknown | 3 | 10.7 |
| MITE | 1 | 3.6 |
| PiggyBac | 1 | 3.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Grande, F.; Jamilloux, V.; Choisne, N.; Calchera, A.; Rolshausen, G.; Petersen, M.; Schulz, M.; Nilsson, M.A.; Schmitt, I. Transposable Elements in the Genome of the Lichen-Forming Fungus Umbilicaria pustulata and Their Distribution in Different Climate Zones along Elevation. Biology 2022, 11, 24. https://doi.org/10.3390/biology11010024
Dal Grande F, Jamilloux V, Choisne N, Calchera A, Rolshausen G, Petersen M, Schulz M, Nilsson MA, Schmitt I. Transposable Elements in the Genome of the Lichen-Forming Fungus Umbilicaria pustulata and Their Distribution in Different Climate Zones along Elevation. Biology. 2022; 11(1):24. https://doi.org/10.3390/biology11010024
Chicago/Turabian StyleDal Grande, Francesco, Véronique Jamilloux, Nathalie Choisne, Anjuli Calchera, Gregor Rolshausen, Malte Petersen, Meike Schulz, Maria A. Nilsson, and Imke Schmitt. 2022. "Transposable Elements in the Genome of the Lichen-Forming Fungus Umbilicaria pustulata and Their Distribution in Different Climate Zones along Elevation" Biology 11, no. 1: 24. https://doi.org/10.3390/biology11010024
APA StyleDal Grande, F., Jamilloux, V., Choisne, N., Calchera, A., Rolshausen, G., Petersen, M., Schulz, M., Nilsson, M. A., & Schmitt, I. (2022). Transposable Elements in the Genome of the Lichen-Forming Fungus Umbilicaria pustulata and Their Distribution in Different Climate Zones along Elevation. Biology, 11(1), 24. https://doi.org/10.3390/biology11010024