
Quantitative Insights into the Contribution of Nematocysts to the Adaptive Success of Cnidarians Based on Proteomic Analysis

 , ,
, ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Collecting Details
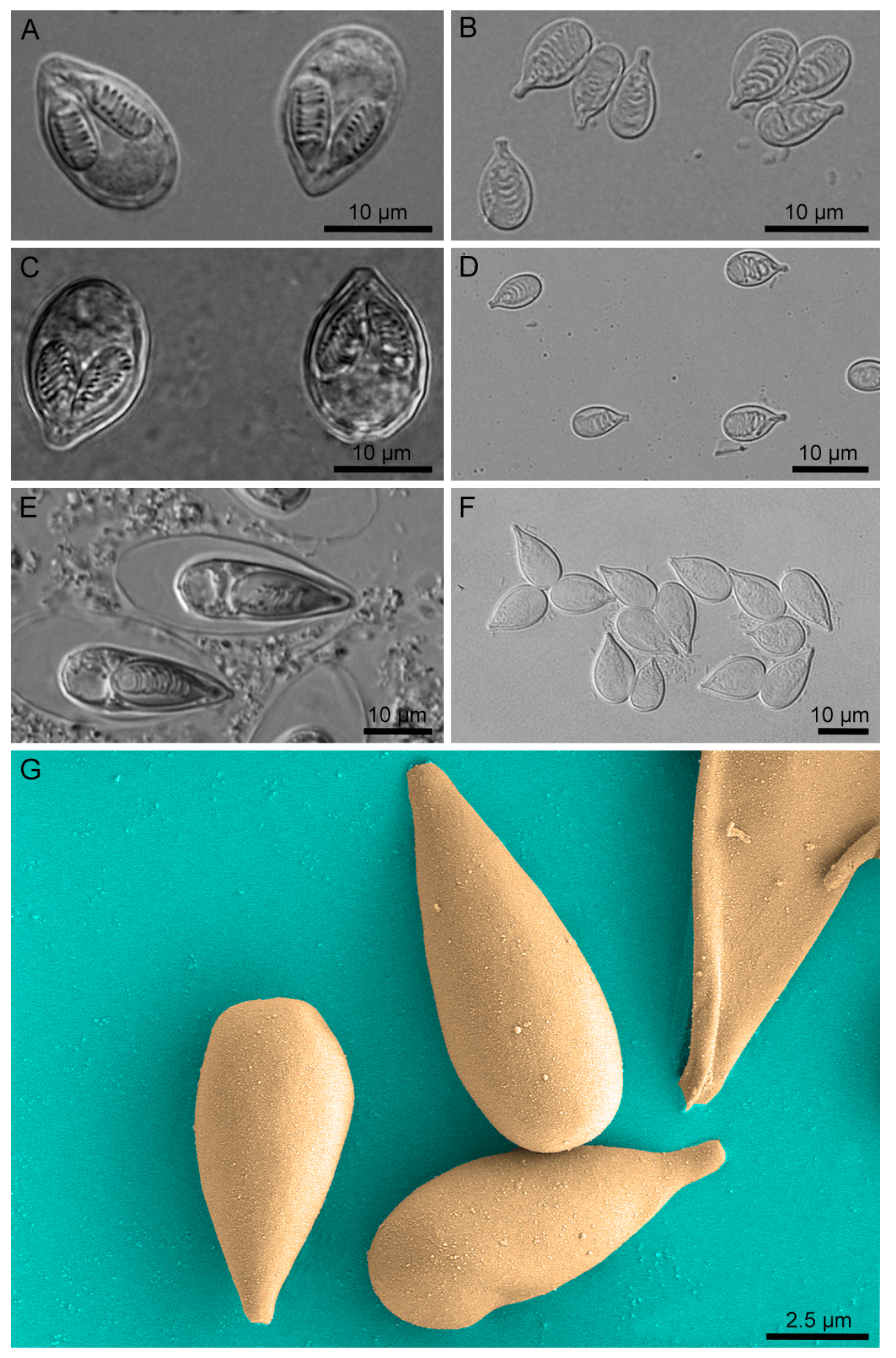
2.2. Nematocyst Isolation and Purity/Integrity Verification
2.3. Next-Generation Sequencing and Assembly
2.4. Data Decontamination and Construction Databases for MS/MS Analysis
2.5. Tandem Mass Spectrometry and Proteomic Analysis
2.6. Comparative Proteomic and Venomic Analyses
2.7. Ortholog Assessment, Gene Selection, and Data Filtering
2.8. Phylogenetic Analysis
2.9. Divergence Time Estimation
2.10. Selection Analyses and Adaptation Quantification
3. Results
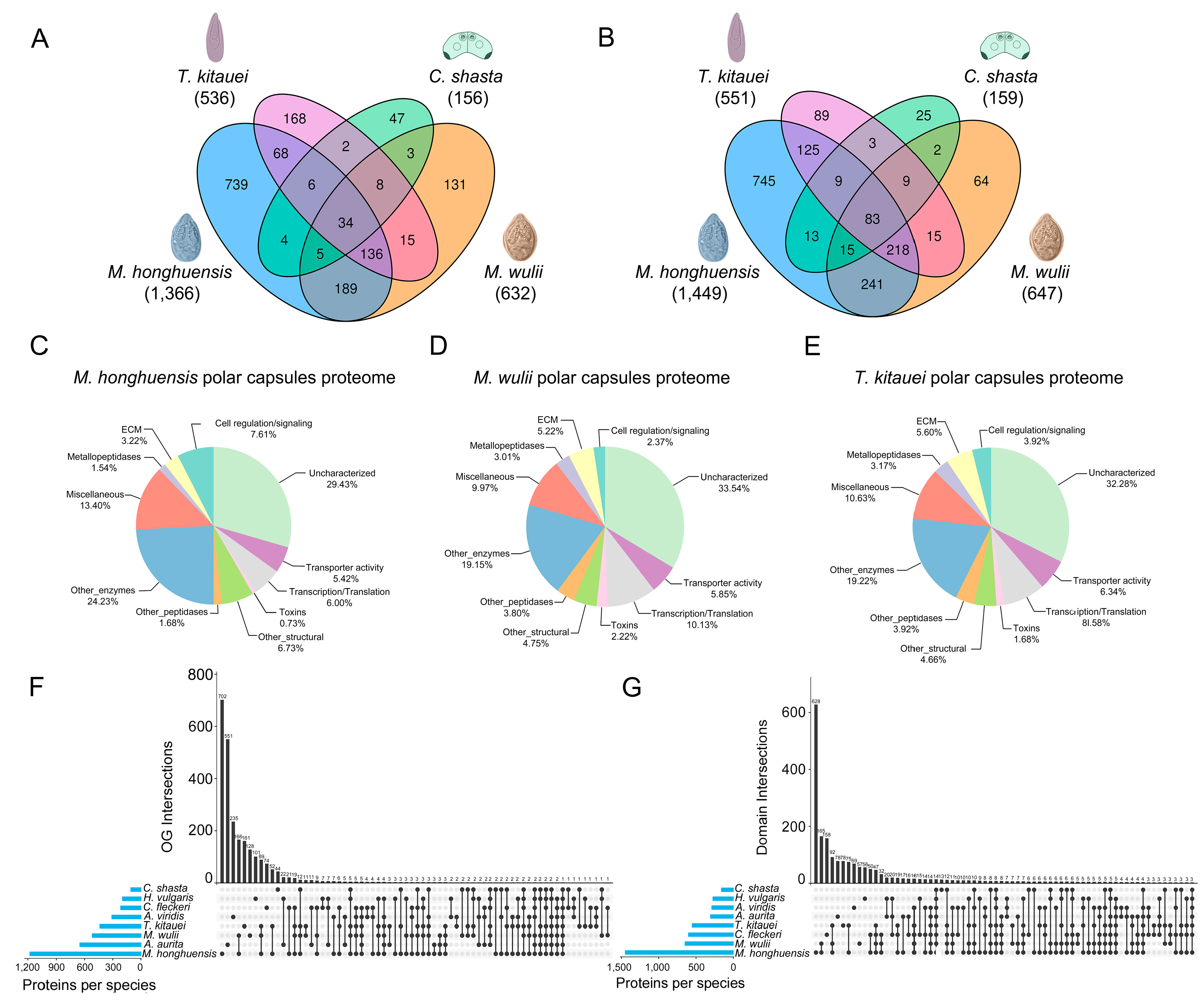
3.1. Comparative Proteomics
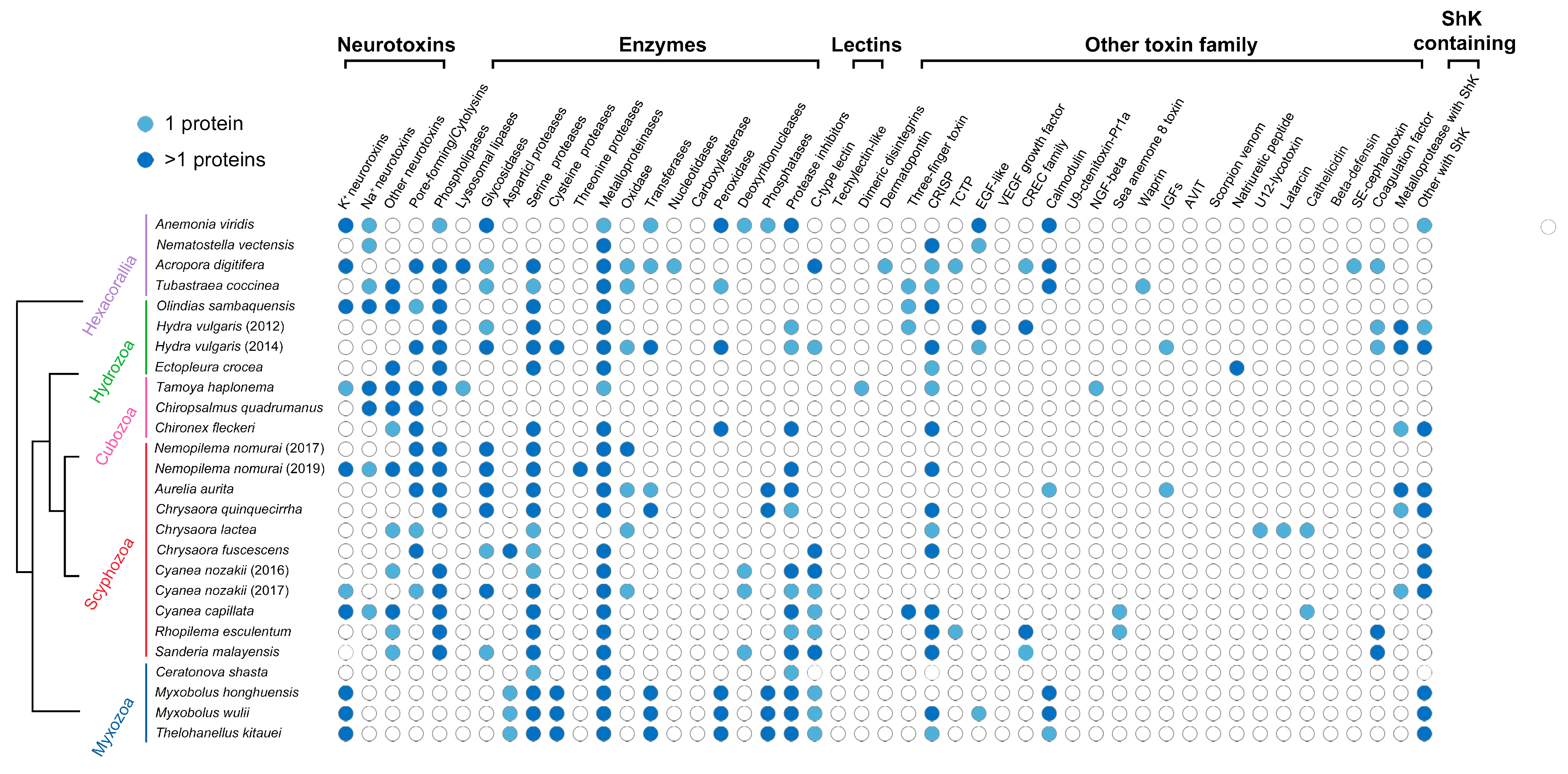
3.2. Myxozoan Venom Analysis
3.3. NEM Core Set
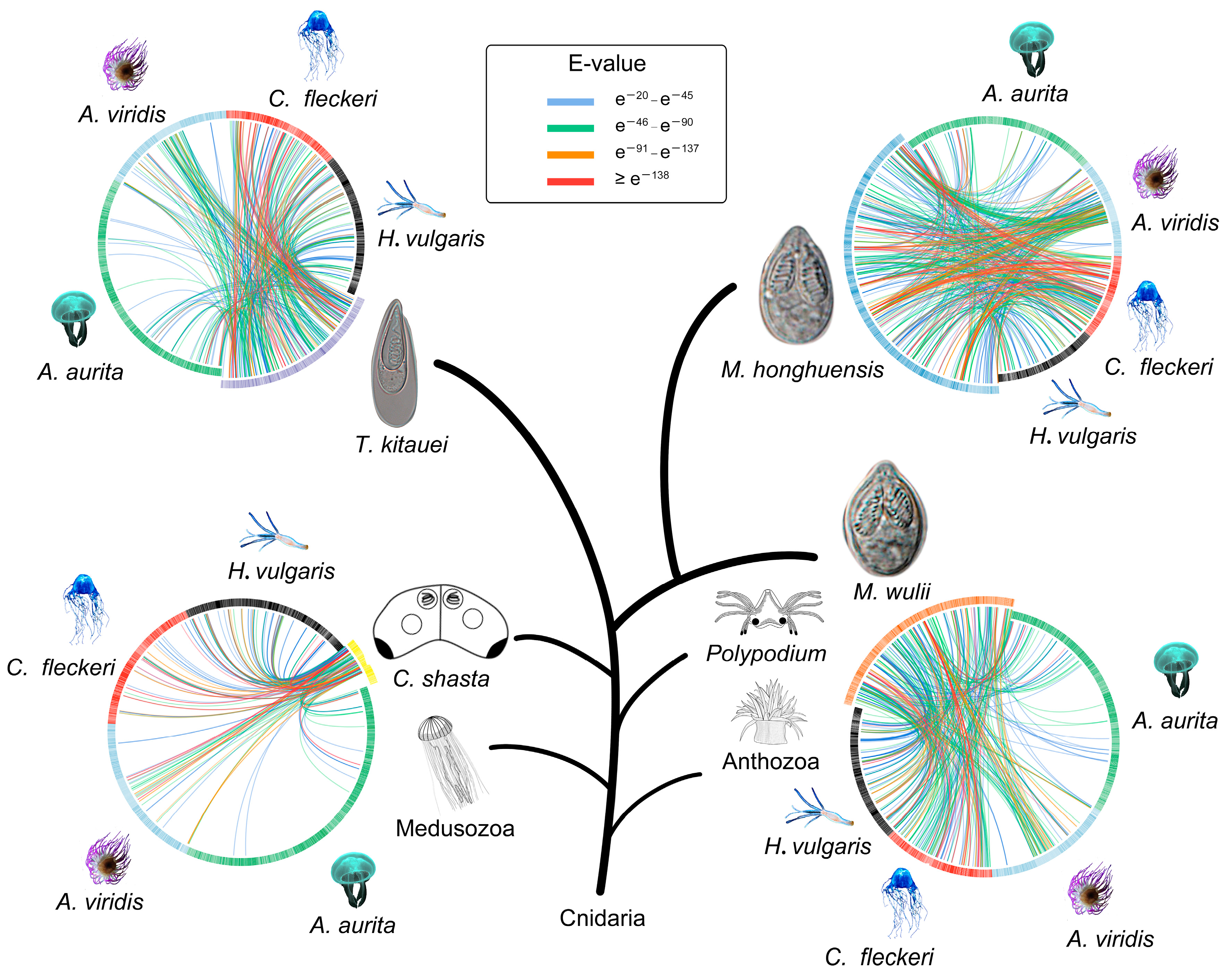
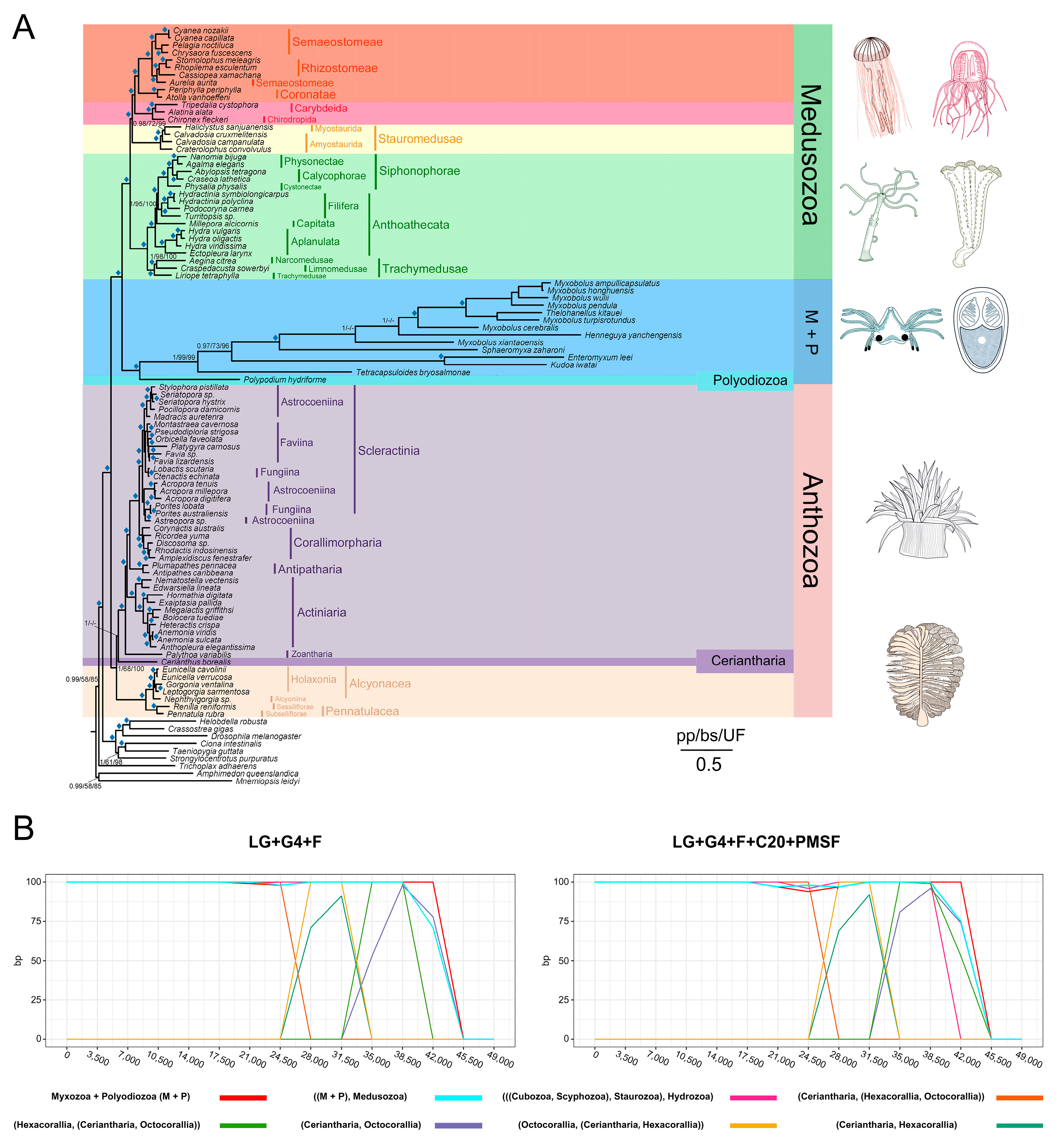
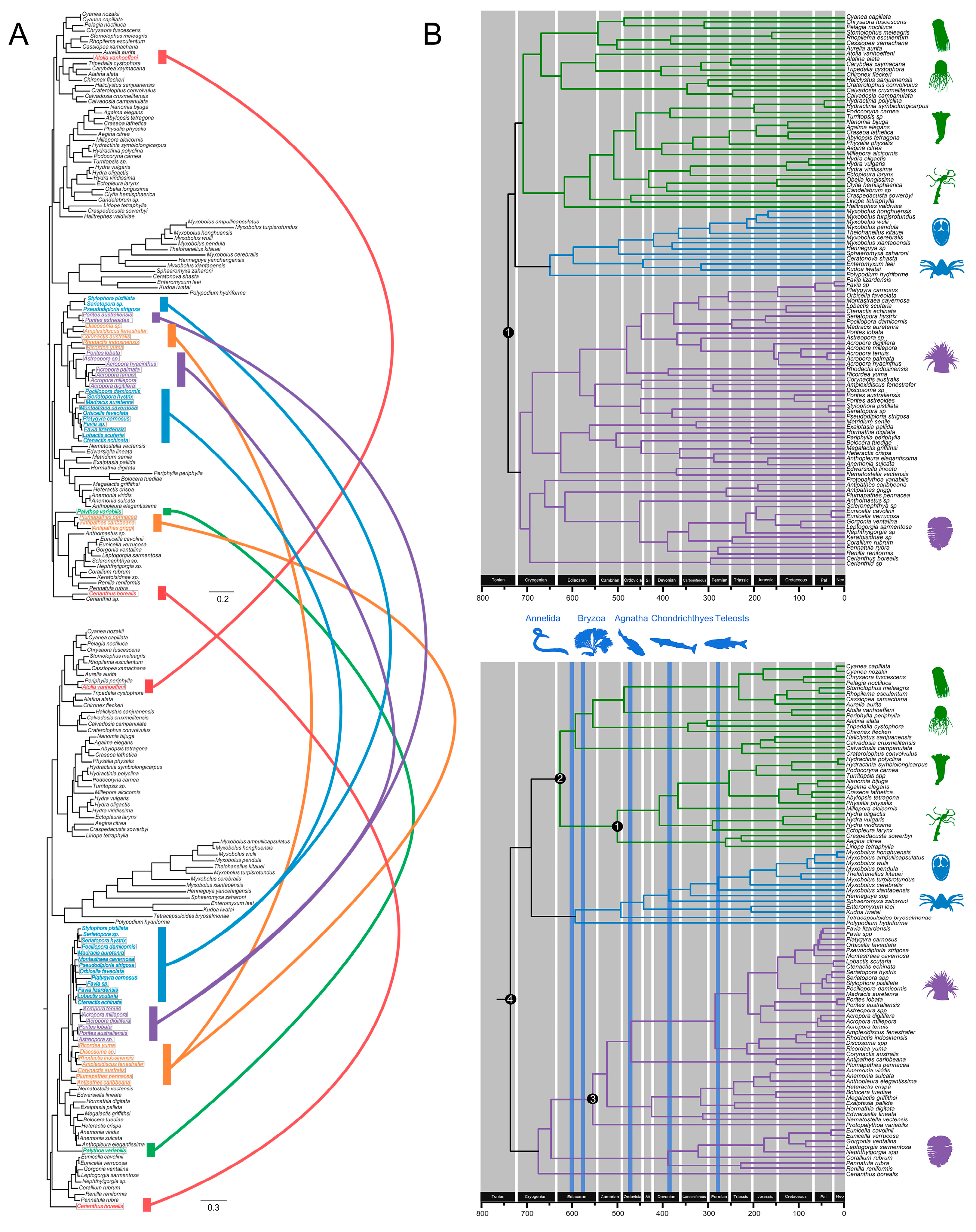
3.4. Cnidarian Organismal and Nematocyst Phylogenetic Analyses
3.5. Major Events in Nematocyst Evolution
3.6. Divergence Time Estimates
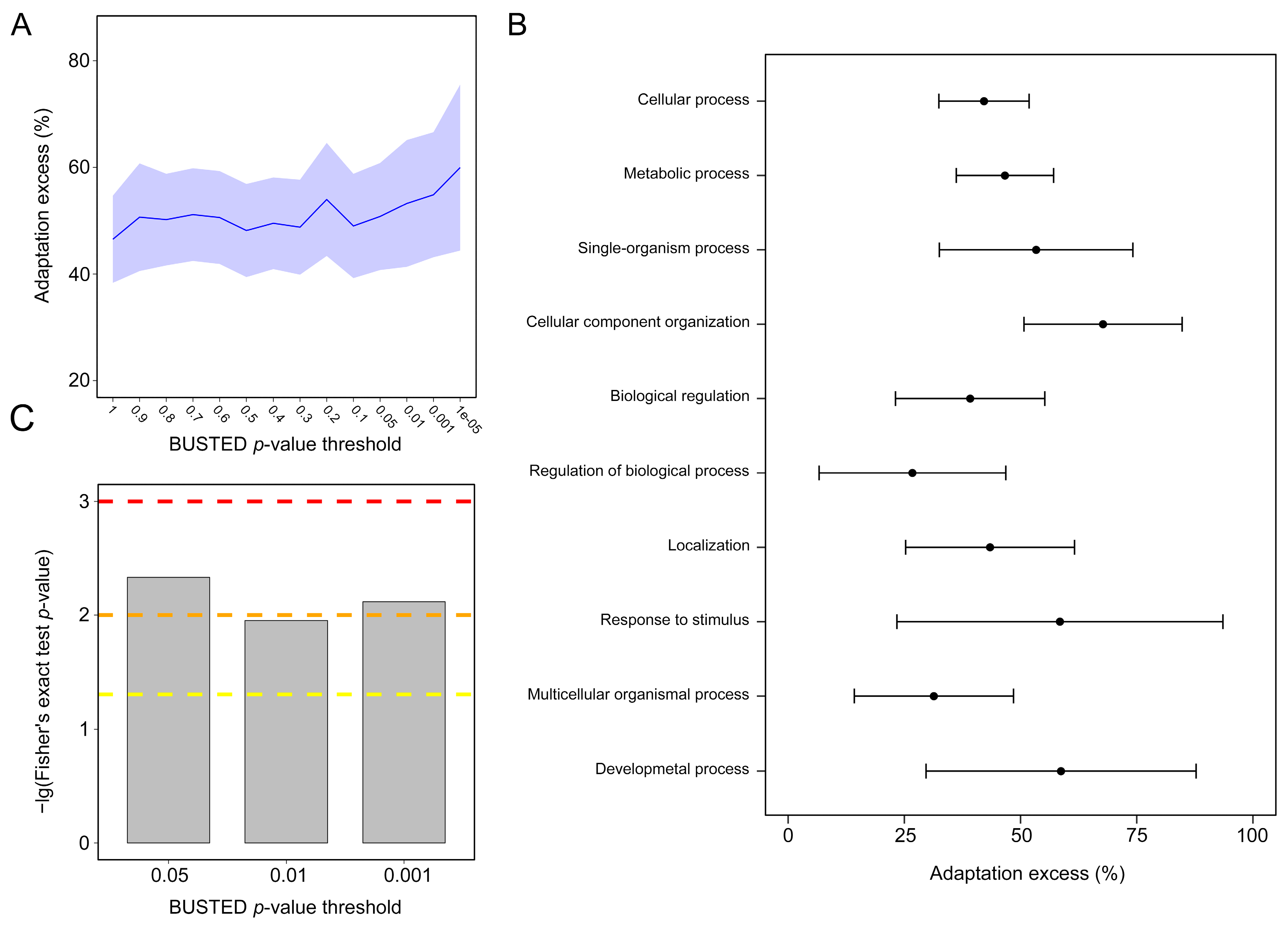
3.7. NEM and Non-NEM Selection and Adaptation Analyses
4. Discussion
4.1. Nematocyst Heterogeneity and Organelle–Organism Discordance Support Extensive Species-Specific Adaptation
4.2. Dynamic Evolution of Myxozoan Venom and Episodic Positive Selection
4.3. Defining the NEM Core Set
4.4. Decentralized Evolutionary Pattern of Nematocysts
4.5. Macroevolutionary Lag between the Nematocyst Core Set and Cnidarian Diversification
4.6. Frequent Adaptation in NEMs across Cnidarians
4.7. Future Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cartwright, P.; Halgedahl, S.L.; Hendricks, J.R.; Jarrard, R.D.; Marques, A.C.; Collins, A.G.; Lieberman, B.S. Exceptionally Preserved Jellyfishes from the Middle Cambrian. PLoS ONE 2007, 2, e1121. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Agrawal, S.; Aranda, M.; Baumgarten, S.; Belcaid, M.; Drake, J.L.; Erwin, D.; Foret, S.; Gates, R.D.; Gruber, D.F. Comparative Genomics Explains the Evolutionary Success of Reef-Forming Corals. Elife 2016, 5, e13288. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, S.D.; Bartholomew, J.L.; Lotan, T. Myxozoans: Ancient Metazoan Parasites Find a Home in Phylum Cnidaria. Zoology 2018, 129, 66–68. [Google Scholar] [CrossRef]
- Hallett, S.L.; Hartigan, A.; Atkinson, S.D. Myxozoans on the Move: Dispersal Modes, Exotic Species and Emerging Diseases. In Myxozoan Evolution, Ecology and Development; Okamura, B., Gruhl, A., Bartholomew, J.L., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 343–362. ISBN 978-3-319-14753-6. [Google Scholar]
- Holzer, A.S.; Bartošová-Sojková, P.; Born-Torrijos, A.; Lövy, A.; Hartigan, A.; Fiala, I. The Joint Evolution of the Myxozoa and Their Alternate Hosts: A Cnidarian Recipe for Success and Vast Biodiversity. Mol. Ecol. 2018, 27, 1651–1666. [Google Scholar] [CrossRef]
- Lotan, A.; Fishman, L.; Loya, Y.; Zlotkin, E. Delivery of a Nematocyst Toxin. Nature 1995, 375, 456. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, A.; Özbek, S. The Nematocyst: A Molecular Map of the Cnidarian Stinging Organelle. Int. J. Dev. Biol. 2012, 56, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Young, G.A.; Hagadorn, J.W. The Fossil Record of Cnidarian Medusae. Palaeoworld 2010, 19, 212–221. [Google Scholar] [CrossRef]
- David, C.N.; Özbek, S.; Adamczyk, P.; Meier, S.; Pauly, B.; Chapman, J.; Hwang, J.S.; Gojobori, T.; Holstein, T.W. Evolution of Complex Structures: Minicollagens Shape the Cnidarian Nematocyst. Trends Genet. 2008, 24, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Nüchter, T.; Benoit, M.; Engel, U.; Özbek, S.; Holstein, T.W. Nanosecond-Scale Kinetics of Nematocyst Discharge. Curr. Biol. 2006, 16, R316–R318. [Google Scholar] [CrossRef] [Green Version]
- Amreen Nisa, S.; Vinu, D.; Krupakar, P.; Govindaraju, K.; Sharma, D.; Vivek, R. Jellyfish Venom Proteins and Their Pharmacological Potentials: A Review. Int. J. Biol. Macromol. 2021, 176, 424–436. [Google Scholar] [CrossRef]
- Kallert, D.M.; Ponader, S.; Eszterbauer, E.; El-Matbouli, M.; Haas, W. Myxozoan Transmission via Actinospores: New Insights into Mechanisms and Adaptations for Host Invasion. Parasitology 2007, 134, 1741–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roop, J.I.; Chang, K.C.; Brem, R.B. Polygenic Evolution of a Sugar Specialization Trade-off in Yeast. Nature 2016, 530, 336. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhou, Y.; Pinto, C.M.; Charles-Dominique, P.; Galindo-González, J.; Zhang, S.; Zhang, J. Evolution of the Sweet Taste Receptor Gene Tas1r2 in Bats. Mol. Biol. Evol. 2010, 27, 2642–2650. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, P.C.; Smith, W.L.; Price, S.A.; Tang, K.L.; Sparks, J.S.; Ferry, L.A.; Kuhn, K.L.; Eytan, R.I.; Near, T.J. The Evolution of Pharyngognathy: A Phylogenetic and Functional Appraisal of the Pharyngeal Jaw Key Innovation in Labroid Fishes and Beyond. Syst. Biol. 2012, 61, 1001–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, C.S.; Eastwood, R.J.; Miotto, S.T.; Hughes, C.E. Multiple Continental Radiations and Correlates of Diversification in Lupinus (Leguminosae): Testing for Key Innovation with Incomplete Taxon Sampling. Syst. Biol. 2012, 61, 443–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cracraft, J. The Origin of Evolutionary Novelties: Pattern and Process at Different Hierarchical Levels. Evol. Innov. 1990, 21–44. [Google Scholar] [CrossRef]
- Balasubramanian, P.G.; Beckmann, A.; Warnken, U.; Schnolzer, M.; Schuler, A.; Bornberg-Bauer, E.; Holstein, T.W.; Ozbek, S. Proteome of Hydra Nematocyst. J. Biol. Chem. 2012, 287, 9672–9681. [Google Scholar] [CrossRef] [Green Version]
- Moran, Y.; Praher, D.; Schlesinger, A.; Ayalon, A.; Tal, Y.; Technau, U. Analysis of Soluble Protein Contents from the Nematocysts of a Model Sea Anemone Sheds Light on Venom Evolution. Mar. Biotechnol. 2013, 15, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Gacesa, R.; Chung, R.; Dunn, S.R.; Weston, A.J.; Jaimes-Becerra, A.; Marques, A.C.; Morandini, A.C.; Hranueli, D.; Starcevic, A.; Ward, M.; et al. Gene Duplications Are Extensive and Contribute Significantly to the Toxic Proteome of Nematocysts Isolated from Acropora Digitifera (Cnidaria: Anthozoa: Scleractinia). BMC Genom. 2015, 16, 774. [Google Scholar] [CrossRef] [Green Version]
- Rachamim, T.; Morgenstern, D.; Aharonovich, D.; Brekhman, V.; Lotan, T.; Sher, D. The Dynamically Evolving Nematocyst Content of an Anthozoan, a Scyphozoan, and a Hydrozoan. Mol. Biol. Evol. 2015, 32, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Jaimes-Becerra, A.; Gacesa, R.; Doonan, L.B.; Hartigan, A.; Marques, A.C.; Okamura, B.; Long, P.F. “Beyond Primary Sequence”—Proteomic Data Reveal Complex Toxins in Cnidarian Venoms. Integr. Comp. Biol. 2019, 59, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Weston, A.J.; Chung, R.; Dunlap, W.C.; Morandini, A.C.; Marques, A.C.; Moura-da-Silva, A.M.; Ward, M.; Padilla, G.; da Silva, L.F.; Andreakis, N.; et al. Proteomic Characterisation of Toxins Isolated from Nematocysts of the South Atlantic Jellyfish Olindias Sambaquiensis. Toxicon 2013, 71, 11–17. [Google Scholar] [CrossRef]
- Brinkman, D.L.; Jia, X.; Potriquet, J.; Kumar, D.; Dash, D.; Kvaskoff, D.; Mulvenna, J. Transcriptome and Venom Proteome of the Box Jellyfish Chironex Fleckeri. BMC Genom. 2015, 16, 407. [Google Scholar] [CrossRef] [Green Version]
- Jaimes-Becerra, A.; Chung, R.; Morandini, A.C.; Weston, A.J.; Padilla, G.; Gacesa, R.; Ward, M.; Long, P.F.; Marques, A.C. Comparative Proteomics Reveals Recruitment Patterns of Some Protein Families in the Venoms of Cnidaria. Toxicon 2017, 137, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, H.; Yue, Y.; Liu, S.; Xing, R.; Chen, X.; Li, P. Combined Proteomics and Transcriptomics Identifies Sting-Related Toxins of Jellyfish Cyanea Nozakii. J. Proteom. 2016, 148, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ponce, D.; Brinkman, D.; Potriquet, J.; Mulvenna, J. Tentacle Transcriptome and Venom Proteome of the Pacific Sea Nettle, Chrysaora Fuscescens (Cnidaria: Scyphozoa). Toxins 2016, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Y.; Yu, H.; Li, R.; Xing, R.; Liu, S.; Li, K.; Wang, X.; Chen, X.; Li, P. Functional Elucidation of Nemopilema Nomurai and Cyanea Nozakii Nematocyst Venoms’ Lytic Activity Using Mass Spectrometry and Zymography. Toxins 2017, 9, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce-Garcia, D.P. Transcriptomic, Proteomic and Biological Analyses of Venom Proteins from Two Chrysaora Jellyfish. Ph.D. Thesis, The University of Melbourne, Melbourne, Australia, 15 June 2017. [Google Scholar]
- Wang, C.; Wang, B.; Wang, B.; Wang, Q.; Liu, G.; Wang, T.; He, Q.; Zhang, L. Unique Diversity of Sting-Related Toxins Based on Transcriptomic and Proteomic Analysis of the Jellyfish Cyanea Capillata and Nemopilema Nomurai (Cnidaria: Scyphozoa). J. Proteome Res. 2019, 18, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.C.N.; Qu, Z.; Nong, W.; Hui, J.H.L.; Ngai, S.M. Proteomic Analysis of the Venom of Jellyfishes Rhopilema Esculentum and Sanderia Malayensis. Mar. Drugs 2020, 18, 655. [Google Scholar] [CrossRef]
- Piriatinskiy, G.; Atkinson, S.D.; Park, S.; Morgenstern, D.; Brekhman, V.; Yossifon, G.; Bartholomew, J.L.; Lotan, T. Functional and Proteomic Analysis of Ceratonova Shasta (Cnidaria: Myxozoa) Polar Capsules Reveals Adaptations to Parasitism. Sci. Rep. 2017, 7, 9010. [Google Scholar] [CrossRef] [Green Version]
- Gavelis, G.S.; Wakeman, K.C.; Tillmann, U.; Ripken, C.; Mitarai, S.; Herranz, M.; Özbek, S.; Holstein, T.; Keeling, P.J.; Leander, B.S. Microbial Arms Race: Ballistic “Nematocysts” in Dinoflagellates Represent a New Extreme in Organelle Complexity. Sci. Adv. 2017, 3, e1602552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lom, J.; Dyková, I. Myxozoan Genera: Definition and Notes on Taxonomy, Life-Cycle Terminology and Pathogenic Species. Folia Parasitol. (Praha) 2013, 53, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.S.; Neuhof, M.; Rubinstein, N.D.; Diamant, A.; Philippe, H.; Huchon, D.; Cartwright, P. Genomic Insights into the Evolutionary Origin of Myxozoa within Cnidaria. Proc. Natl. Acad. Sci. USA 2015, 112, 14912–14917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, B.; Gruhl, A. Evolution, Origins and Diversification of Parasitic Cnidarians. In The Evolution and Fossil Record of Parasitism: Identification and Macroevolution of Parasites; De Baets, K., Huntley, J.W., Eds.; Topics in Geobiology; Springer International Publishing: Cham, Switzerland, 2021; pp. 109–152. ISBN 978-3-030-42484-8. [Google Scholar]
- Stilwell, J.M.; Griffin, M.J.; Rosser, T.G.; Mohammed, H.H.; Sidor, I.F.; Camus, A.C. Insights into Myxozoan Composition and Physiology Revealed by Histochemical Properties of Myxospores. J. Fish Dis. 2020, 43, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Americus, B.; Austin, B.M.; Lotan, T.; Bartholomew, J.L.; Atkinson, S.D. In Vitro and In Vivo Assays Reveal That Cations Affect Nematocyst Discharge in Myxobolus cerebralis (Cnidaria: Myxozoa). Parasitology 2020, 147, 1352–1358. [Google Scholar] [CrossRef]
- Cannon, Q.; Wagner, E. Comparison of Discharge Mechanisms of Cnidarian Cnidae and Myxozoan Polar Capsules. Rev. Fish. Sci. 2003, 11, 185–219. [Google Scholar] [CrossRef]
- Ben-David, J.; Atkinson, S.D.; Pollak, Y.; Yossifon, G.; Shavit, U.; Bartholomew, J.L.; Lotan, T. Myxozoan Polar Tubules Display Structural and Functional Variation. Parasit. Vectors 2016, 9, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Americus, B.; Lotan, T.; Bartholomew, J.L.; Atkinson, S.D. A Comparison of the Structure and Function of Nematocysts in Free-Living and Parasitic Cnidarians (Myxozoa). Int. J. Parasitol. 2020, 50, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Naldoni, J.; Zatti, S.A.; da Silva, M.R.M.; Maia, A.A.M.; Adriano, E.A. Morphological, Ultrastructural, and Phylogenetic Analysis of Two Novel Myxobolus Species (Cnidaria: Myxosporea) Parasitizing Bryconid Fish from São Francisco River, Brazil. Parasitol. Int. 2019, 71, 27–36. [Google Scholar] [CrossRef]
- Lom, J.; Arthur, J.R. A Guideline for the Preparation of Species Descriptions in Myxosporea. J. Fish Dis. 1989, 12, 151–156. [Google Scholar] [CrossRef]
- Council, N.R. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2010. [Google Scholar]
- Guo, Q.; Liu, Y.; Zhai, Y.; Gu, Z. A Fast and Effective Method for Dissecting Parasitic Spores: Myxozoans as an Example. J. Exp. Biol. 2020, 223, jeb214916. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices Clustering Tools (TGICL): A Software System for Fast Clustering of Large EST Datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Guo, Q.; Li, D.; Zhai, Y.; Gu, Z. CCPRD: A Novel Analytical Framework for the Comprehensive Proteomic Reference Database Construction of Nonmodel Organisms. ACS Omega 2020, 5, 15370–15384. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of Genome Assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Lomsadze, A.; Burns, P.D.; Borodovsky, M. Integration of Mapped RNA-Seq Reads into Automatic Training of Eukaryotic Gene Finding Algorithm. Nucl. Acids Res. 2014, 42, e119. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A Web Server for Gene Prediction in Eukaryotes That Allows User-Defined Constraints. Nucl. Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [Green Version]
- Korf, I. Gene Finding in Novel Genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D. Improving the Arabidopsis Genome Annotation Using Maximal Transcript Alignment Assemblies. Nucl. Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of Protein Coding Regions in RNA Transcripts. Nucl. Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473, 337. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M. InterPro in 2017—Beyond Protein Family and Domain Annotations. Nucl. Acids Res. 2016, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A Package for the Generation of Highly-Customizable Venn and Euler Diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Darzentas, N. Circoletto: Visualizing Sequence Similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Gacesa, R.; Barlow, D.J.; Long, P.F. Machine Learning Can Differentiate Venom Toxins from Other Proteins Having Non-Toxic Physiological Functions. Peer J. Comput. Sci. 2016, 2, e90. [Google Scholar] [CrossRef]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot Program: A Central Hub of Integrated Venom Protein Data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebersberger, I.; Strauss, S.; von Haeseler, A. HaMStR: Profile Hidden Markov Model Based Search for Orthologs in ESTs. BMC Evol. Biol. 2009, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Cannon, J.T.; Vellutini, B.C.; Smith, J.; Ronquist, F.; Jondelius, U.; Hejnol, A. Xenacoelomorpha Is the Sister Group to Nephrozoa. Nature 2016, 530, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kocot, K.M.; Citarella, M.R.; Moroz, L.L.; Halanych, K.M. PhyloTreePruner: A Phylogenetic Tree-Based Approach for Selection of Orthologous Sequences for Phylogenomics. Evol. Bioinform. 2013, 9, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Struck, T.H. TreSpEx-Detection of Misleading Signal in Phylogenetic Reconstructions Based on Tree Information. Evol. Bioinform. 2014, 10, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucl. Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Roure, B.; Rodriguez-Ezpeleta, N.; Philippe, H. SCaFoS: A Tool for Selection, Concatenation and Fusion of Sequences for Phylogenomics. BMC Evol. Biol. 2007, 7, S2. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian Software Package for Phylogenetic Reconstruction and Molecular Dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimodaira, H. An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.W.; Sharpe, S.C.; Silberman, J.D.; Heiss, A.A.; Lang, B.F.; Simpson, A.G.; Roger, A.J. Phylogenomics Demonstrates That Breviate Flagellates Are Related to Opisthokonts and Apusomonads. Proc. R. Soc. Lond. B Biol. Sci. 2013, 280, 20131755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susko, E.; Field, C.; Blouin, C.; Roger, A.J. Estimation of Rates-across-Sites Distributions in Phylogenetic Substitution Models. Syst. Biol. 2003, 52, 594–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, M.J. R8s: Inferring Absolute Rates of Molecular Evolution and Divergence Times in the Absence of a Molecular Clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Hwang, D.-S.; Lee, J.-S.; Song, J.-I.; Seo, T.-K.; Won, Y.-J. Estimation of Divergence Times in Cnidarian Evolution Based on Mitochondrial Protein-Coding Genes and the Fossil Record. Mol. Phylogenet. Evol. 2012, 62, 329–345. [Google Scholar] [CrossRef]
- Schwentner, M.; Bosch, T.C.G. Revisiting the Age, Evolutionary History and Species Level Diversity of the Genus Hydra (Cnidaria: Hydrozoa). Mol. Phylogenet. Evol. 2015, 91, 41–55. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Shinzato, C.; Shoguchi, E.; Kawashima, T.; Hamada, M.; Hisata, K.; Tanaka, M.; Fujie, M.; Fujiwara, M.; Koyanagi, R.; Ikuta, T.; et al. Using the Acropora Digitifera Genome to Understand Coral Responses to Environmental Change. Nature 2011, 476, 320–323. [Google Scholar] [CrossRef]
- Chapman, J.A.; Kirkness, E.F.; Simakov, O.; Hampson, S.E.; Mitros, T.; Weinmaier, T.; Rattei, T.; Balasubramanian, P.G.; Borman, J.; Busam, D.; et al. The Dynamic Genome of Hydra. Nature 2010, 464, 592–596. [Google Scholar] [CrossRef]
- Prada, C.; Hanna, B.; Budd, A.F.; Woodley, C.M.; Schmutz, J.; Grimwood, J.; Iglesias-Prieto, R.; Pandolfi, J.M.; Levitan, D.; Johnson, K.G.; et al. Empty Niches after Extinctions Increase Population Sizes of Modern Corals. Curr. Biol. 2016, 26, 3190–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voolstra, C.R.; Li, Y.; Liew, Y.J.; Baumgarten, S.; Zoccola, D.; Flot, J.-F.; Tambutté, S.; Allemand, D.; Aranda, M. Comparative Analysis of the Genomes of Stylophora Pistillata and Acropora Digitifera Provides Evidence for Extensive Differences between Species of Corals. Sci. Rep. 2017, 7, 17583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Xiong, J.; Zhou, Z.; Huo, F.; Miao, W.; Ran, C.; Liu, Y.; Zhang, J.; Feng, J.; Wang, M.; et al. The Genome of the Myxosporean Thelohanellus Kitauei Shows Adaptations to Nutrient Acquisition within Its Fish Host. Genome Biol. Evol. 2014, 6, 3182–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgarten, S.; Simakov, O.; Esherick, L.Y.; Liew, Y.J.; Lehnert, E.M.; Michell, C.T.; Li, Y.; Hambleton, E.A.; Guse, A.; Oates, M.E.; et al. The Genome of Aiptasia, a Sea Anemone Model for Coral Symbiosis. Proc. Natl. Acad. Sci. USA 2015, 112, 11893–11898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putnam, N.H.; Srivastava, M.; Hellsten, U.; Dirks, B.; Chapman, J.; Salamov, A.; Terry, A.; Shapiro, H.; Lindquist, E.; Kapitonov, V.V. Sea Anemone Genome Reveals Ancestral Eumetazoan Gene Repertoire and Genomic Organization. Science 2007, 317, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enard, D.; Cai, L.; Gwennap, C.; Petrov, D.A. Viruses Are a Dominant Driver of Protein Adaptation in Mammals. Elife 2016, 5, e12469. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Sasikumar, A.N.; Perez, W.B.; Kinzy, T.G. The Many Roles of the Eukaryotic Elongation Factor 1 Complex. Wiley Interdiscip. Rev. RNA 2012, 3, 543–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbert, P.B.; Henikoff, S. Histone Variants—Ancient Wrap Artists of the Epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Whitley, D.; Goldberg, S.P.; Jordan, W.D. Heat Shock Proteins: A Review of the Molecular Chaperones. J. Vasc. Surg. 1999, 29, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.S.; Takaku, Y.; Momose, T.; Adamczyk, P.; Özbek, S.; Ikeo, K.; Khalturin, K.; Hemmrich, G.; Bosch, T.C.; Holstein, T.W.; et al. Nematogalectin, a Nematocyst Protein with GlyXY and Galectin Domains, Demonstrates Nematocyte-Specific Alternative Splicing in Hydra. Proc. Natl. Acad. Sci. USA 2010, 107, 18539–18544. [Google Scholar] [CrossRef] [Green Version]
- Draizen, E.J.; Shaytan, A.K.; Mariño-Ramírez, L.; Talbert, P.B.; Landsman, D.; Panchenko, A.R. HistoneDB 2.0: A Histone Database with Variants—An Integrated Resource to Explore Histones and Their Variants. Database 2016, 2016, baw014. [Google Scholar] [CrossRef] [Green Version]
- Kayal, E.; Bentlage, B.; Sabrina Pankey, M.; Ohdera, A.H.; Medina, M.; Plachetzki, D.C.; Collins, A.G.; Ryan, J.F. Phylogenomics Provides a Robust Topology of the Major Cnidarian Lineages and Insights on the Origins of Key Organismal Traits. BMC Evol. Biol. 2018, 18, 68. [Google Scholar] [CrossRef] [Green Version]
- Ashwood, L.M.; Norton, R.S.; Undheim, E.A.B.; Hurwood, D.A.; Prentis, P.J. Characterising Functional Venom Profiles of Anthozoans and Medusozoans within Their Ecological Context. Mar. Drugs 2020, 18, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouiaei, M.; Yanagihara, A.A.; Madio, B.; Nevalainen, T.J.; Alewood, P.F.; Fry, B.G. Ancient Venom Systems: A Review on Cnidaria Toxins. Toxins 2015, 7, 2251–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambra, I.; Lauritano, C. A Review of Toxins from Cnidaria. Mar. Drugs 2020, 18, 507. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Moran, Y. The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals. PLoS Genet. 2015, 11, e1005596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klompen, A.M.L.; Kayal, E.; Collins, A.G.; Cartwright, P. Phylogenetic and Selection Analysis of an Expanded Family of Putatively Pore-Forming Jellyfish Toxins (Cnidaria: Medusozoa). Genome Biol. Evol. 2021, 13, evab081. [Google Scholar] [CrossRef] [PubMed]
- Shpirer, E.; Chang, E.S.; Diamant, A.; Rubinstein, N.; Cartwright, P.; Huchon, D. Diversity and Evolution of Myxozoan Minicollagens and Nematogalectins. BMC Evol. Biol. 2014, 14, 205. [Google Scholar] [CrossRef] [Green Version]
- Kyslík, J.; Kosakyan, A.; Nenarokov, S.; Holzer, A.S.; Fiala, I. The Myxozoan Minicollagen Gene Repertoire Was Not Simplified by the Parasitic Lifestyle: Computational Identification of a Novel Myxozoan Minicollagen Gene. BMC Genom. 2021, 22, 198. [Google Scholar] [CrossRef]
- Ganot, P.; Moya, A.; Magnone, V.; Allemand, D.; Furla, P.; Sabourault, C. Adaptations to Endosymbiosis in a Cnidarian-Dinoflagellate Association: Differential Gene Expression and Specific Gene Duplications. PLoS Genet. 2011, 7, e1002187. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Ochman, H. Stepwise Formation of the Bacterial Flagellar System. Proc. Natl. Acad. Sci. USA 2007, 104, 7116–7121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arivalagan, J.; Yarra, T.; Marie, B.; Sleight, V.A.; Duvernois-Berthet, E.; Clark, M.S.; Marie, A.; Berland, S. Insights from the Shell Proteome: Biomineralization to Adaptation. Mol. Biol. Evol. 2017, 34, 66–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abby, S.S.; Rocha, E.P.C. The Non-Flagellar Type III Secretion System Evolved from the Bacterial Flagellum and Diversified into Host-Cell Adapted Systems. PLoS Genet. 2012, 8, e1002983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogales, M.; Padrón, B.; Padilla, D.P.; Nieves, C.; Marrero, P. Germination Patterns throughout an Insular Altitudinal Gradient: The Case of the Macaronesian Endemic Plant Rubia Fruticosa Ait. (Rubiaceae) in El Hierro (Canary Islands). Flora-Morphol. Distrib. Funct. Ecol. Plants 2009, 204, 543–548. [Google Scholar] [CrossRef]
- Cartwright, P.; Collins, A. Fossils and Phylogenies: Integrating Multiple Lines of Evidence to Investigate the Origin of Early Major Metazoan Lineages. Integr. Comp. Biol. 2007, 47, 744–751. [Google Scholar] [CrossRef]
- Peterson, K.J.; Lyons, J.B.; Nowak, K.S.; Takacs, C.M.; Wargo, M.J.; McPeek, M.A. Estimating Metazoan Divergence Times with a Molecular Clock. Proc. Natl. Acad. Sci. USA 2004, 101, 6536–6541. [Google Scholar] [CrossRef] [Green Version]
- Waggoner, B.; Collins, A.G. Reductio Ad Absurdum: Testing the Evolutionary Relationships of Ediacaran and Paleozoic Problematic Fossils Using Molecular Divergence Dates. J. Paleontol. 2004, 78, 51–61. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [Green Version]
- Turing, A.M. The Chemical Basis of Morphogenesis. Bull. Math. Biol. 1990, 52, 153–197. [Google Scholar] [CrossRef]
- Von Neumann, N.; Burks, A.W. Theory of Self-Reproducing Automata; University of Illinois Press: Champaign, IL, USA, 1966; ISBN 978-0-598-37798-2. [Google Scholar]
- Gierer, A.; Berking, S.; Bode, H.; David, C.N.; Flick, K.; Hansmann, G.; Schaller, H.; Trenkner, E. Regeneration of Hydra from Reaggregated Cells. Nat. New Biol. 1972, 239, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinhardt, H. Turing’s Theory of Morphogenesis of 1952 and the Subsequent Discovery of the Crucial Role of Local Self-Enhancement and Long-Range Inhibition. Interface Focus 2012, 2, 407–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Jin, L.; Zhang, N.; Petridis, A.K.; Eckert, T.; Scheiner-Bobis, G.; Bergmann, M.; Scheidig, A.; Schauer, R.; Yan, M.; et al. The Sialic Acid-Dependent Nematocyst Discharge Process in Relation to Its Physical-Chemical Properties Is a Role Model for Nanomedical Diagnostic and Therapeutic Tools. Mar. Drugs 2019, 17, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucl. Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | M. honghuensis ID | M. wulii ID | T. kitauei ID |
|---|---|---|---|
| Elongation factor 1 alpha 1 | 75931_MH_MC | 23790_WL_MC | 165085_TK_MC |
| Canonical histone H2A | 32284_MH_MC | 49939_WL_MC | 80662_TK_MC |
| Canonical histone H2B | 28864_MH_MC | 51458_WL_MC | 62843_TK_MC |
| Heat shock cognate 71 kDa (Hsc70) | 8642_MH_MC | 159445_WL_MC | 85197_TK_MC |
| Nematogalectin-related | 75931_MH_MC | 23790_WL_MC | 165085_TK_MC |
| Matrix_Name | Constraint_Tree_Iqtree | Log L | Δlog L | p-Value AU | Bootstrap RELL |
|---|---|---|---|---|---|
| 4_103tx_146og | Best_iqtree_ML | −2,303,638.671 | 0.000 | 0.6220 | 0.4661 |
| Best_Bayes | −2,303,688.375 | 49.704 | 0.1477 | 0.1261 | |
| [Polypodiozoa + Hydrozoa] | −2,304,312.129 | 673.458 | 0.0001 | 0.0000 | |
| [Polypodiozoa + Aegina_citrea] | −2,305,903.793 | 2265.122 | 0.0018 | 0.0000 | |
| [[[Myxo + Polypodiozoa] + Anthozoa] + Medusozoa] | −2,304,295.467 | 656.796 | 0.0004 | 0.0000 | |
| [[Medusozoa + Octocorallia] + Hexacorallia] | −2,305,100.274 | 1461.603 | 0.0025 | 0.0000 | |
| [[[Ceriantharia+ Hexacorallia] + Octocorallia] + Medusozoa] | −2,303,639.715 | 1.044 | 0.5813 | 0.4078 | |
| [[[Cubozoa+ Scyphozoa] + Hydrozoa] + Staurozoa] | −2,303,758.459 | 119.787 | 0.0001 | 0.0000 | |
| [[Staurozoa + Cubozoa] + [Hydrozoa + Scyphozoa]] | −2,304,131.216 | 492.544 | 0.0000 | 0.0000 |
| Present Study | Holzer et al. 2018 | Kodádková et al. 2015 | Park et al. 2012 | Waggoner & Collins 2004 | Cartwright & Collins 2007 | Medina et al. 2006 | Peterson et al. 2004 | |
|---|---|---|---|---|---|---|---|---|
| Markers | 146 single-copy nuclear orthologs | 6 nuclear protein-coding genes | 18S rRNA gene | 13 mitochondrial protein-coding genes | 18S rRNA gene | 18S and 28S rRNA genes | 13 mitochondrial protein-coding genes | 7 nuclear protein-coding genes |
| Node age | ||||||||
| Deep nodes | ||||||||
| Cnidaria | 736.2 | 786 (723–848) | - | 741 (686–819) | 875 (700–1200) | 711.7 (389, 1035) | - | 595 (561–626) |
| “M + P” | 592.9 | 725 (667–779) | - | - | - | - | - | - |
| Medusozoa | 626.6 | - | - | 607 (571–670) | 790 (640–940) | - | - | - |
| “M +P” + Medusozoa | 689.6 | - | - | - | - | - | - | - |
| Shallow nodes | ||||||||
| P. hydriforme | 592.9 | 651 (601–700) | - | - | - | - | - | - |
| Myxozoa | 492.6 | 588 (540–642) | 530 (457–603) | - | - | - | - | - |
| Cubozoa | 344.5 | - | - | - | - | - | - | - |
| Scyphozoa | 485.3 | - | - | - | - | - | - | - |
| Cubozoa + Scyphozoa | 554.2 | - | - | - | - | 537.5 (191–943) | - | - |
| Staurozoa | 225.5 | - | - | - | - | - | - | - |
| Octocorallia | 388.5 | - | - | 292 (173, 421) | - | - | - | - |
| Hexacorallia | 554.4 | - | - | 601 (544, 684) | - | 517.5 | - | - |
| Scleractinia + Cora | 284.3 | - | - | 276 (235–323) | - | - | 264 (240–280) | - |
| Cora | 214.2 | - | - | 206 (158–253) | - | - | 121 (110–132) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Q.; Whipps, C.M.; Zhai, Y.; Li, D.; Gu, Z. Quantitative Insights into the Contribution of Nematocysts to the Adaptive Success of Cnidarians Based on Proteomic Analysis. Biology 2022, 11, 91. https://doi.org/10.3390/biology11010091
Guo Q, Whipps CM, Zhai Y, Li D, Gu Z. Quantitative Insights into the Contribution of Nematocysts to the Adaptive Success of Cnidarians Based on Proteomic Analysis. Biology. 2022; 11(1):91. https://doi.org/10.3390/biology11010091
Chicago/Turabian StyleGuo, Qingxiang, Christopher M. Whipps, Yanhua Zhai, Dan Li, and Zemao Gu. 2022. "Quantitative Insights into the Contribution of Nematocysts to the Adaptive Success of Cnidarians Based on Proteomic Analysis" Biology 11, no. 1: 91. https://doi.org/10.3390/biology11010091