1. Introduction
Galloway–Mowat syndrome (GAMOS; OMIM 251300) is a rare recessive genetic disease characterized by neurodevelopmental defects and progressive renal glomerulopathy. Neurological involvement of GAMOS contains microcephaly, developmental delay, intellectual disability, and other variable neural symptoms [
1]. Renal manifestations range from isolated proteinuria to early nephrotic syndrome (NS), which may rapidly progress to end-stage renal disease. The prognosis of GAMOS is poor, and the majority of affected children died before six years of age due to therapy-resistant renal failure [
2,
3].
To date, mutations in 11 different genes, such as WDR73, KEOPS complex genes, YRDC, and NUP107, have been reported to cause GAMOS [
4,
5,
6,
7]. Although WDR73 is the first gene to be identified in GAMOS, the molecular mechanisms underlying the pathophysiology of GAMOS upon WDR73 function remain obscure. WDR73 encodes a WD40-repeat-containing protein that can be expressed in a variety of cells. In the fetal kidney, WDR73 exists in immature podocytes from the S-shaped body stage to the capillary-loop stage. In mature glomeruli, WDR73 has a punctate distribution at the periphery of the glomerular tuft and is present in the cell bodies of mature podocytes. WDR73 is localized in the cytoplasm during interphase and accumulates at spindle poles and microtubule asters during mitosis; thereby, it may play a role in cellular architecture and cell cycle [
4]. It is reported that WDR73 could interact with α- and β-tubulin in fibroblasts [
8]. Loss of WDR73 also has been demonstrated to disrupt the integrator complex, perturbing the transcription of cell cycle regulatory proteins [
9]. Despite these observations providing some insights into the function of WDR73, the regulatory pathway of WDR73, the precise contributions of WDR73 to cell physiological function, and the mechanisms underlying GAMOS, especially nephrotic syndrome caused by WDR73 deficiency, are poorly understood.
In this study, we knocked out the WDR73 in human embryonic kidney (HEK) 293 cells to observe the morphological characteristics of the cells and elucidate the functions of WDR73. Additionally, we used a combination of proteomics, transcriptomics, and biochemical assays to identify the regulated targets of WDR73. We aimed to discover directly interacting molecules and the regulatory pathway of WDR73 and to illustrate the molecular mechanism between the WDR73 pathway and nephrotic disease in Galloway–Mowat syndrome. From the molecular mechanism we found in vitro, we draw a hypothesis that the damage to focal adhesion of podocytes caused by WDR73 defect is the key issue of kidney dysfunction. Finally, we verified the hypothesis in a Wdr73 CKO mouse model.
2. Materials and Methods
2.1. Cell Culture and Reagents
HEK 293 cells were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C under 5% CO2. Transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions. The following reagents were used in this study: cycloheximide (CHX), MG132, and chloroquine (Selleck, Houston, TX, USA).
2.2. WDR73-Knockout (KO) and Stable Cell Lines
HEK293-WDR73 KO cells were generated using the CRISPR/Cas9 system. Briefly, plasmids carrying sgRNA targeting the WDR73 region encompassing exon 6 were introduced into HEK293 cells using Lipofectamine 3000. Transfected HEK293 colonies were selected using puromycin (Solarbio, Beijing, China). The puromycin-selected colonies were seeded into 96-well plates (1 cell per well) and expanded. Furthermore, WDR73 with frameshift deletions in the six exons was confirmed by Sanger sequencing. Western blotting was used to examine WDR73 protein expression in each clone. WDR73 KO cells were transfected with plasmids carrying WT WDR73 using Lipofectamine 3000 and selected using hygromycin. Cell lines carrying the stably integrated plasmid were expanded from a single colony. The colony with an approximate protein expression level was selected.
2.3. Plasmids and Antibodies
Plasmids were constructed as follows: Plasmids expressing WT WDR73 were generated by cloning human WDR73 into pcDNA3.1-Hygro vector. For co-immunoprecipitation, a plasmid expressing 3×HA-WDR73 was generated by cloning human WDR73 into an in-house modified version of the pcDNA3.1(+) -3×HA vector. PCMV-GFP-hEPG5 was kindly provided by Professor Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences, China). Plasmids expressing GFP were generated by cloning GFP into a PCMA vector. For the GST pulldown assay, PIP4K2C was PCR-amplified and cloned into the pGEX-6P-1 vector to produce GST-tag fused recombinant proteins, whereas WDR73 was PCR-amplified and cloned into the pET28A vector to produce His-tag fused recombinant proteins. Plasmids for WDR73 KO were constructed by inserting sgRNA sequences into the pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid. The sgRNA sequences specifically targeting WDR73 were as follows: F, CACCGACTTCGGAGCCTCGCCCCA; R, AAACTGGGGCGAGGCTCCGAAGTC. The antibodies used in this study are shown in
Table S4.
2.4. Cell Proliferation Assay
The proliferative ability of the cells was measured using the CCK-8 assay (Vazyme, Nanjing, China) according to the manufacturer’s instructions. Briefly, cells were plated into a 96-well plate at a density of 2.0 × 103 cells/well and incubated at 37 °C for 0, 24, 48, 72, and 96 h. Twenty microliters of CCK-8 reagent was added to each well, and the cells were cultured for 2 h. All experiments were performed in triplicates. Absorbance was measured at 450 nm using a microplate reader (ELx800, BioTek, Winooski, VT, USA).
2.5. Cell Cycle and Apoptosis Assays
The cells were harvested after 72 h, and the cell suspension was then digested. Next, the cells were fixed using ethanol (75%) for 4 h at 4 °C, and the supernatant was subsequently discarded, followed by incubation with an RNA enzyme containing iodide (PI, Sigma-Aldrich, St. Louise, MO, USA). After the cells were washed three times with PBS, the cell cycle was detected using a Cytek Dxp Athena flow cytometer (Cytek, Biosciences, San Diego, CA, USA), and data analysis was conducted using the Modfit LT software. For the determination of apoptosis, the cells were stained with FITC-conjugated annexin V and PI according to the manufacturer’s instructions (Vazyme, Nanjing, China). Data were collected and analyzed using a Cytek Dxp Athena flow cytometer, and data analysis was performed using the FlowJo software 10.4 (BD, Ashland, USA). All experiments were performed in triplicates.
2.6. Cell Adhesion and Spreading Assay
For all cell lines, 2.0 × 103 cells/well were seeded onto 96-well plates, and four regions per well were imaged every 2 h over a period of 48 h using the Incucyte Zoom (Essen BioScience, Ann Arbor, MI, USA). The cell areas were measured using the Incucyte Zoom software package to obtain quantitative data on the extent of cell spreading.
2.7. Immunofluorescence
The coverslip-grown cells were fixed in 4% paraformaldehyde for 20 min at room temperature or in ice-cold MeOH for 5 min. The coverslips were permeabilized in 0.5% Triton X-100/PBS for 15 min and then blocked in 5% bovine serum albumin/PBS for 1 h at room temperature. Primary antibodies were applied in a dilution according to the instructions on staining buffer overnight at 4 °C. Secondary antibodies (Alexa Fluor secondary 488; cy3, Jackson, West Grove, PA, USA) were applied in a 1:200 dilution in staining buffer for 1 h at 37 °C in the dark. The nuclei were stained with DAPI. Actin filaments were labeled with FS488 phalloidin (Solarbio, Beijing, China). All images were captured using a confocal laser scanning microscope (Zeiss LSM 880 with airyscan, Zeiss, Berlin, Germany). The primary antibodies used were described previously.
2.8. Human Proteome Microarray Assays
The recombinant His-WDR73 fusion protein was purified by MerryBio Co., Ltd. (Huai’an, China). After quantification and qualification, the recombinant His-WDR73 fusion proteins were labeled with Cy3 (CyDye Protein LabellingCY3 MONO 5-PACK, GE) and used to probe the Arrayit HuProt v4.0 20 K Human Proteome Microarrays (CDI Laboratories, Baltimore, MD, USA). Briefly, Cy3-tagged His-WDR73 was incubated on the microarray overnight at 4 °C and washed. Finally, the microarray was scanned using a microarray scanner (CapitalBio, Beijing, China). Data were analyzed to generate a candidate list of WDR73-binding proteins; the signal-to-noise ratio (SNR) was defined as the ratio of the median foreground value to the median background value. A total of 336 candidates were selected based on a Z-score > 3 (Z-score = (SNR-mean)/SD). Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were conducted to reveal the unique biological significance and key pathways associated with WDR73 in the candidate list of WDR73-binding proteins (criteria: p-value < 0.05, significantly enriched).
2.9. Western Blotting
Cells were harvested and lysed using RIPA lysis buffer (Beyotime, Haimen, China) containing 1 mM PMSF and 1× protease inhibitor cocktail (Sigma-Aldrich, St. Louise, MO, USA). Cell lysates were separated using SDS-PAGE and transferred to a PVDF membrane (Merck Millipore, Burlington, MA, USA). The membranes were blocked in Tris-buffered saline containing 0.01% Tween20 and 5% non-fat milk for 1 h and then incubated with specific primary antibodies. Following incubation with horseradish peroxidase (HRP)-linked secondary antibody (Jackson, West Grove, PA, USA) at room temperature for 1 h, detection was performed using an Immobilon Western Chemiluminescent HRP substrate kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The primary antibodies used were as previously described.
2.10. GST Pulldown Assays
The plasmids encoding GST, GST-PIP4K2C, and His-WDR73 were transfected into E. coli. The fusion proteins were prepared as previously described. Approximately 100 µg of GST and GST-PIP4K2C fusion protein was immobilized in 50 µL of Mag-Beads GST Fusion Protein Purification (Sangon Biotech, Shanghai, China) and equilibrated before being incubated together at 4 °C for 60 min with a gentle rocking motion. Approximately 100 µg of His-WDR73 fusion protein was added to immobilized GST-PIP4K2C and GST after three washes with PBST. The two fusion proteins were incubated overnight at 4 °C with gentle agitation. The bound proteins were washed five times with PBS, boiled with loading buffer for 5 min, and analyzed by Western blotting, as described previously.
2.11. Co-Immunoprecipitation (Co-IP)
HEK293 cells were transfected with plasmids expressing 3HA-WDR73, GFP-Epg5, 3HA-WDR73, and GFP using Lipofectamine 3000 (Invitrogen Waltham, MA, USA). Cells were lysed using immunoprecipitation buffer (20 mM Tris (pH7.5), 150 mM NaCl, 1% Triton X-100, 1 mM PMSF, and 1× protease inhibitor cocktail (Sigma-Aldrich St. Louise, MO, USA) 48 h after transfection. For Co-IP, approximately 1000 μg protein was incubated at 4 °C overnight with 30 μL indicated monoclonal Anti-HA magnetic beads (Bimake, Houston, TX, USA). The precipitates were washed three times with immunoprecipitation buffer, boiled with loading buffer for 5 min, and analyzed by Western blotting, as described previously.
2.12. PI(4,5)P2 ELISA Assay
PIP2 was quantified using a PI(4,5)P2 mass ELISA kit (echelon, K-4500, Salt Lake City, UT, USA). Briefly, lipids were extracted from WDR73 KO and WT cells according to the manufacturer’s instructions. The PIP2 levels were measured according to the manufacturer’s instructions. The absorbance was measured at 450 nm using a microplate reader (ELx800, BioTek, Winooski, VT, USA). Finally, the quantity of PIP2 was calculated using a standard curve.
2.13. RNA Sequencing
Total RNA was extracted from WDR73 KO and WT cells using TRIzol reagent (Invitrogen, Waltham, MA, USA). After RNA quantification and qualification, library preparation and transcriptome sequencing were performed using Novogene Bioinformatics Technology (Beijing, China). Differential expression analysis was performed using the DESeq2 R package. Finally, enrichment analysis of differentially expressed genes (DEGs), GO enrichment analysis, and KEGG pathway analysis were conducted using clusterProfiler (Bioconductor, Boston, MA, USA) to reveal the unique biological significance and key pathways associated with WDR73 in the DEGs (criteria: p-value < 0.05, significantly enriched). The Disease Ontology (DO) database describes the function of human genes and diseases, and we used clusterProfiler software to test the statistical enrichment of DEGs in the DO pathway (criteria: p < 0.05, significantly enriched).
2.14. RNA Isolation and Quantitative Reverse Transcription (qRT-PCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Waltham, MA, USA), and mRNA was reverse transcribed using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The real-time PCR assay was conducted using 2× SYBR Green Mix (Thermo Fisher Scientific, Waltham, MA, USA) on an ASA-9600 qRT-PCR System (Lanzhou Baiyun Gene Technology Co., Ltd, Lanzhou, China). Primers used for real-time PCR are listed in
Tables S2, S3 and S5. Relative expression was calculated using the 2
−ΔΔCt method.
2.15. Mice
Mice were housed in a specific-pathogen-free facility and kept in a 12 h day/night cycle with free access to chow and water. Genotyping and breeding of animals were performed according to standard procedures. Wdr73 general KO mice were generated by Shanghai Biomodel Organisms Center using CRISPR/Cas9. A guide RNA targeting the KO exon 6–exon 8 of the Wdr73 (ENSMUST00000026816.14) gene was designed. The guide RNA1 and guide RNA2 sequences are shown in
Table S5. Male Wdr73 frameshift heterozygous mice and female Wdr73 frameshift heterozygous mice were mated to obtain homozygous mice. For genotype identification, the Wdr73 frameshift mutation was identified by PCR amplification using the primers PI, PII, PIII, and PIV; (
Table S5). All mouse lines were maintained in the C57BL/6J background by regular backcrossing to the C57BL/6J line. Study protocols complied with all relevant ethical regulations and were approved by the IRB of Central South University (IRB: 2019-2-17).
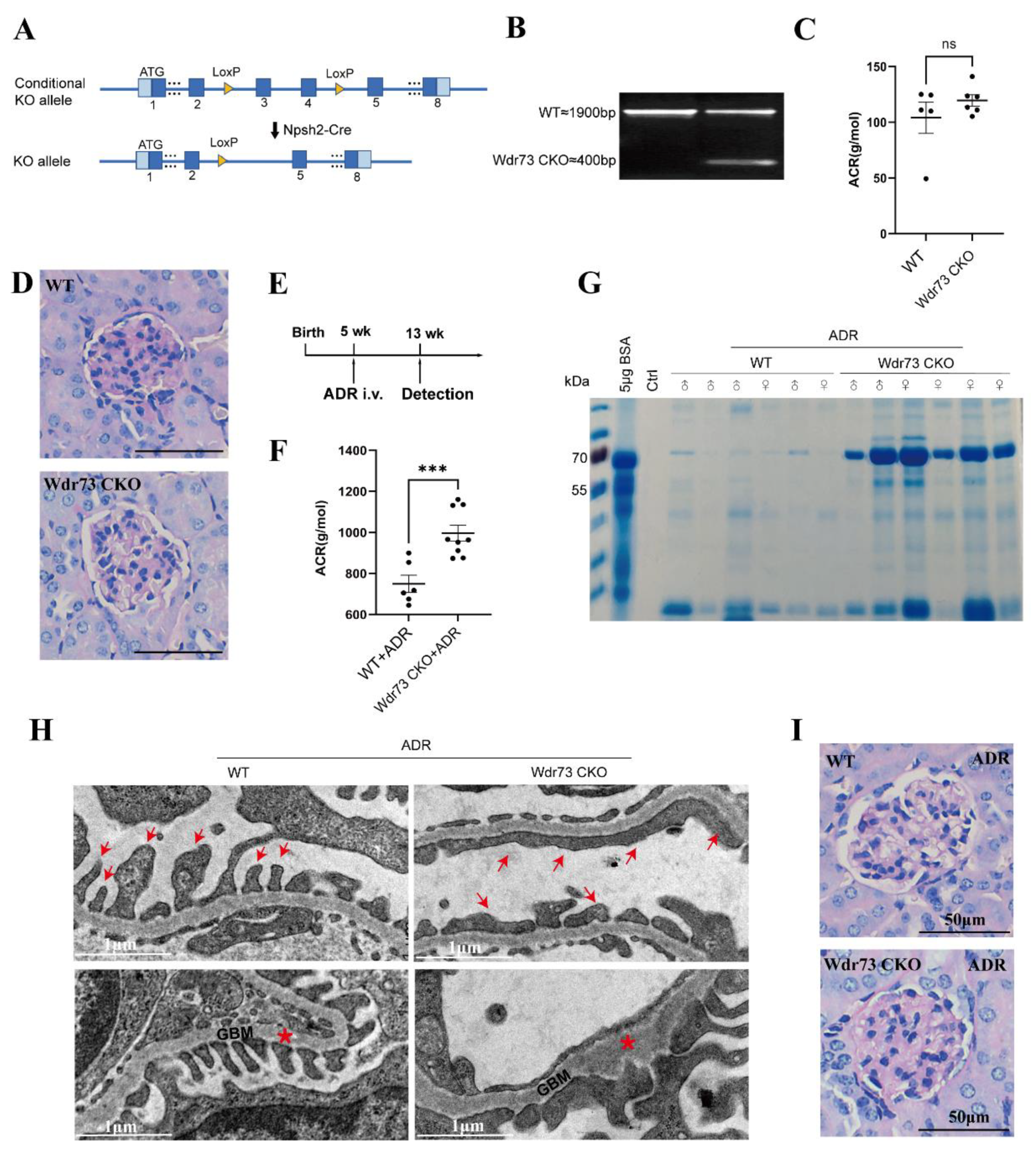
Wdr73 flox mice were generated by GemPharmatech Co., Ltd. (Nanjing, China), using CRISPR/Cas9 on a C57BL/6 background. These mice carry a cassette with LoxP sites flanking a region containing 154 bp coding sequence of Wdr73 exon 4–exon 5, the knocking out of which will result in disruption of protein function. For the generation of a podocyte-specific Wdr73 knockout mouse model, Wdr73flox/flox mice were crossed with Npsh2-Cre mice on a C57BL/6J background (GemPharmatech Co., Ltd, Nanjing, China). In the nephropathy experiments, male and female mice (aged 5–8 weeks), were treated with a single retroorbital injection of ADR (doxorubicin HCl; Macklin, shanghai, China) at the dose of 20 mg/kg for Nphs2-Cre, Wdr73flox/flox (Wdr73 CKO), and Wdr73flox/flox (WT) mice. For genotype identification, the Wdr73 flox was identified by PCR amplification using the primers Wdr73-5F/R and Wdr73-3F/R; Cre recombinase was identified by PCR amplification using the primers Nphs2-cre F/R (
Table S5). General PCR amplification was performed to verify Wdr73 gene knockout efficiency using the primers Wdr73-5F and Wdr73-3R (
Table S5).
The age of animals used for the respective experiments is stated in the figures and/or figure legends (male and female animals showed similar phenotypes and were combined for the analysis).
2.16. Measurement of Urinary Albumin and Creatinine
Albumin and creatinine levels were quantified by measuring spot urine from Wdr73 CKO and WT mice at defined time points. Proteinuria was expressed as albumin–creatinine ratio. Assessment of urinary albumin was performed using a mouse-specific, albumin fluorescence-based kit (Fankewei, Shanghai, China). Measurement of creatinine was performed using an enzymatic creatinine kit (Fankewei, Shanghai, China).
2.17. Urine Analysis for Albumin by SDS–PAGE
First, 16 μL of collected spot urine was mixed with 4 μL 4 × LDS Sample Buffer (NP0007, Invitrogen, Waltham, MA, USA); the sample was heated at 70 °C for 10 min for optimal results and separated by SDS–PAGE. Gels were stained with Coomassie for 1 h and destained using standard methods.
2.18. Renal Ultrastructural Analysis
Fresh kidneys underwent primary fixation with 2% glutaraldehyde in PBS. They were then postfixed in 1% osmium tetroxide for 1 h and dehydrated in 50%, 70%, 90%, 95%, or 100% ethanol and propylene oxide for 10 min each. Samples were further infiltrated with an epoxy resin mixture. Ultrathin sections were collected on copper grids, and sections were stained using 10% uranyl acetate in 50% methanol and modified Sato lead stain. A HITACHI HT7700 electron microscope was used for picture acquisition (Lab of Biomedical Electronic Microscopy Higher Research Center, Central South University, Changsha, China).
2.19. Histology Staining of Kidney Sections
Fresh kidney immersion was fixed in 4% PFA in phosphate-buffered saline (PBS) for 24 h and subsequently dehydrated in 20% or 30% sucrose solution for 24 h each at 4 °C. Kidney cryosections of O.C.T.-embedded (20 μm thick) (Sakura TissueTek #4583, Sakura Finetek, Alphen aan den Rijn, the Netherlands) and kidney sections (3–4 μm thick) of paraffin-embedded (FFPE) tissue were generated using standard methods. FFPE sections were deparaffinized and rehydrated and then underwent heat-induced antigen treatment.
2.20. Mice Glomerular Isolation and Podocyte Culture
Glomeruli were isolated from male or female mice aged between 5 and 8 weeks old using previously described methods [
10]. Mice are anesthetized with Avertin (250 mg/kg body weight) and perfused via the left heart ventricle with 20–40 mL PBS. Isolated kidneys were minced in ice PBS and digested with collagenase type II (2 mg/mL; C5138, Sigma-Aldrich St. Louise, MO, USA) at 37 °C for 30 min, and glomeruli were sequentially sieved with a 100 μm and 70 μm cell strainer in order and washed with PBS. The isolated glomeruli were seeded on collagen I (C8062, Solarbio, Beijing, China)-coated plates. The method yields primary podocytes with 90% purity, confirmed via staining with the podocyte-specific marker nephrin on day 24 after isolation. Primary mouse podocytes were cultured in RPMI medium (Gibco, Waltham, MA, USA) supplemented with 10% FBS and 1% penicillin–streptomycin (Thermo Fisher Scientific Waltham, MA, USA) at 37 °C under 5% CO2, and they were fed with fresh medium every 2–3 days.
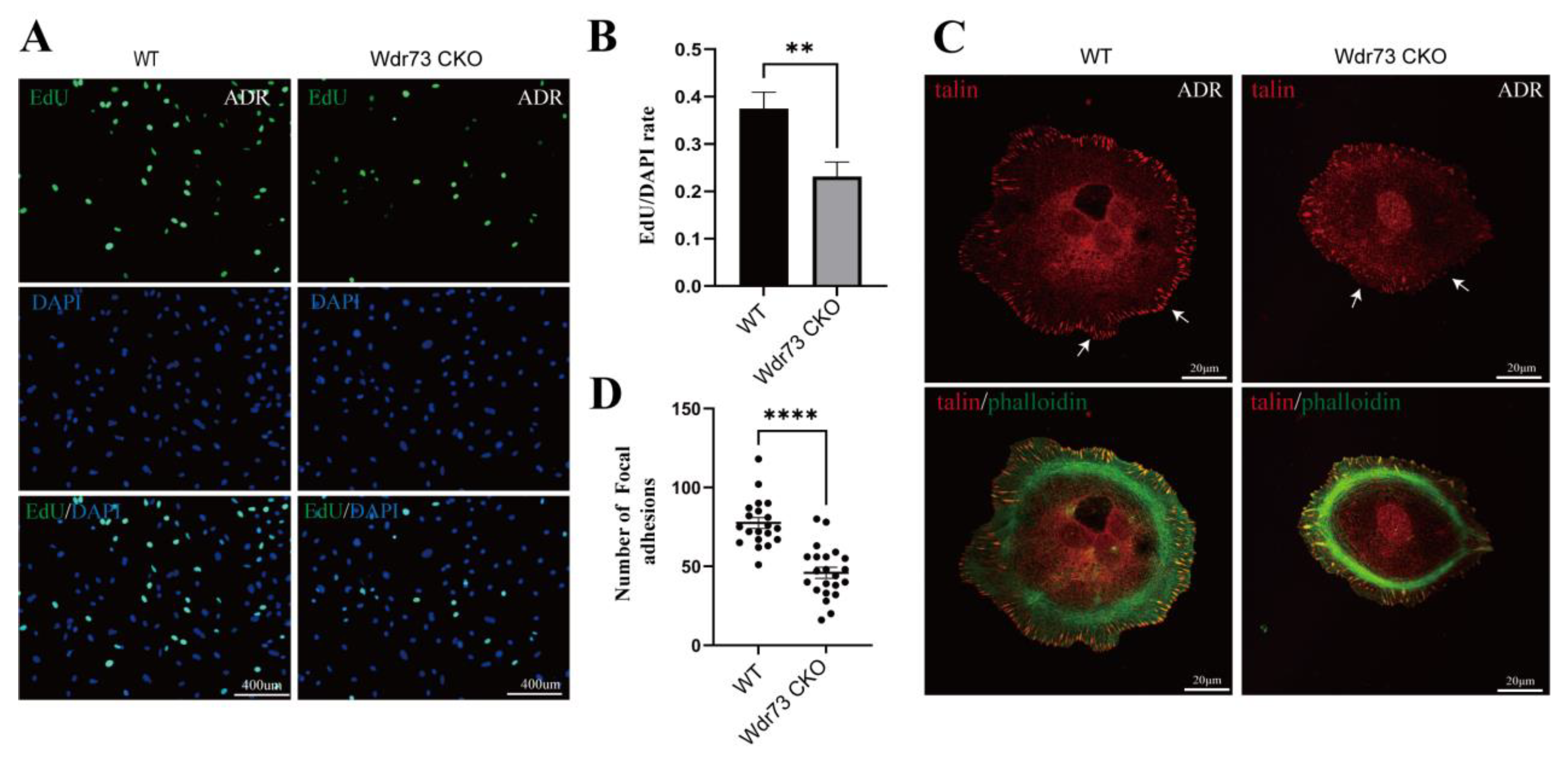
2.21. EdU Assay of Primary Podocytes
Podocyte proliferation was measured using the EdU assay kit (C0071S, Beyotime, Haimen, China). The podocytes were fixed with 0.5% buffered paraformaldehyde and then incubated with the EdU detection solution for 30 min in the dark. Hoechst (C0071S, Beyotime, Haimen, China) was applied to the podocytes, which were incubated in the dark for 10 min. Finally, cell proliferation was analyzed by fluorescence microscopy (Zeiss LSM 880 with airyscan, Zeiss, Berlin, Germany).
4. Discussion
In this study, we revealed that WDR73 exerts an essential function in the regulation of focal adhesion and the actin cytoskeleton and provides a novel molecular mechanism in reduced renal cell proliferation, adhesion, and spreading. By human protein microarray assay and GST pulldown, we confirmed that WDR73 can directly interact with PIP4K2C, which usually heterodimerizes with PIP4K2A and catalyzes the phosphorylation of phosphatidylinositol 5-phosphate (PI5P) to generate PIP2. WDR73 depletion led to a significant reduction in the PIP4K2C and PIP4K2A proteins, consequently inducing a dramatic decrease in intracellular PIP2.
PIP2 plays a key role in FA assembly and actin polymerization [
18]. FA complexes provide the main sites for cell adhesion to the ECM and are associated with the actin cytoskeleton, which is required for cell adhesion and spindle orientation in cell division [
14,
22,
23]. Actually, FA and ECM alterations in podocytes have been widely reported to be one of the main cellular bases for proteinuria. FA is a key signal and structural hub of foot processes that regulate the actin cytoskeleton, through which podocytes could firmly bind to the GBM, which is a dense matrix of ECM components. Once the FA dynamic or assembly is disrupted, podocytes gradually detach from the GBM, leading to the irreversible progression of kidney disease [
24,
25,
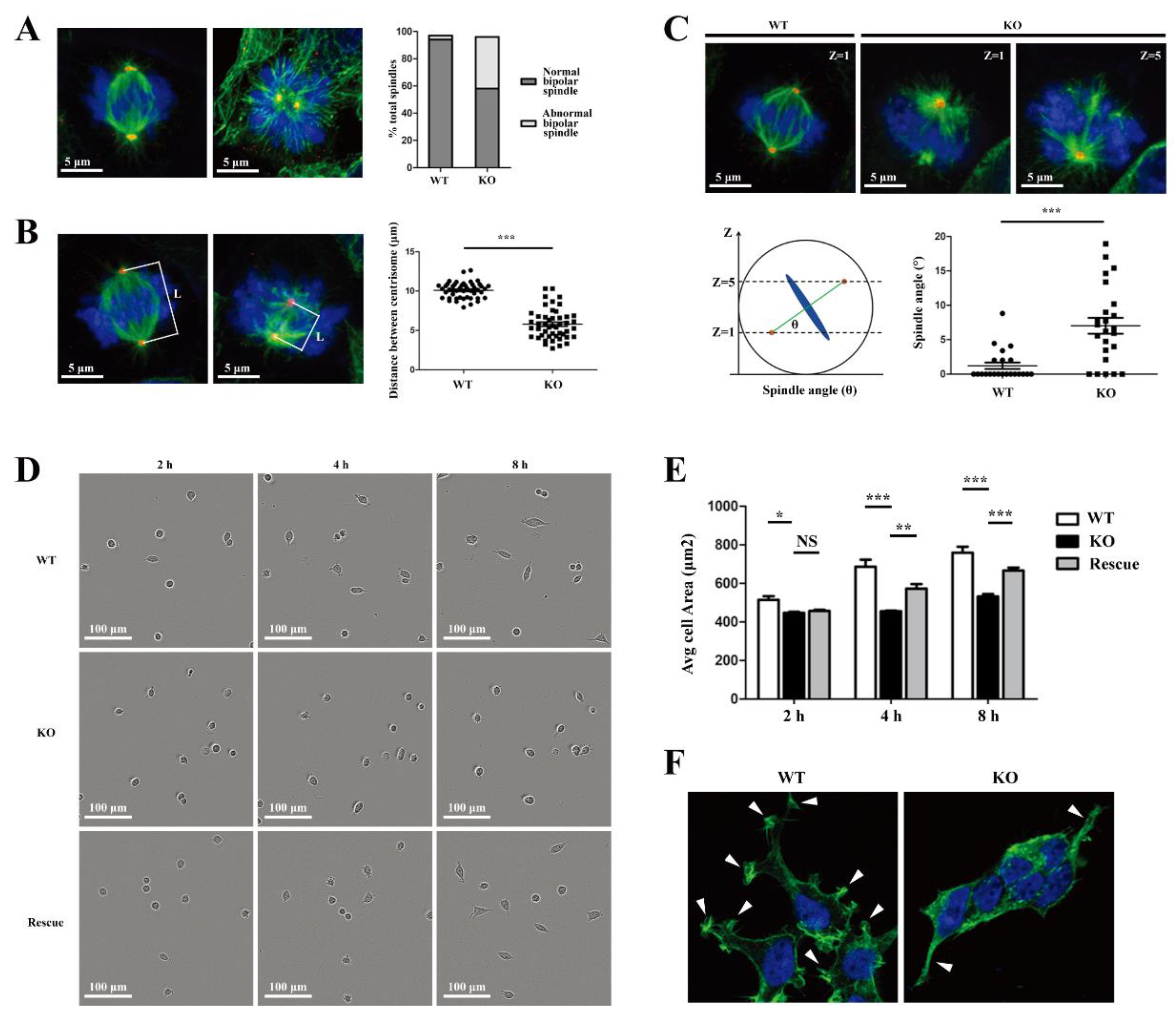
26]. We demonstrated that there were significant changes in cell morphology of WDR73 KO cells, with significantly reduced cell spreading area and less pseudopod formation. The adhesion ability also decreased dramatically. We observed that the number of speckle-like adhesion spots was significantly reduced and the shape was ambiguous in WDR73 KO cells. Considering the RNA-seq results, the enrichment of DEGs in FA and ECM-related pathways together, these results suggest that FA and actin filament assembly were disrupted by the absence of WDR73, leading to pseudopod formation, cell adhesion, and spreading abnormality, which may be the primary cellular pathological basis for the albuminuria and other renal abnormalities in GAMOS1. Although this point has not been mentioned in other studies on GAMOS, we confirmed that the FA formation and distribution were impaired in primary podocytes derived from ADR-injected Wdr73 CKO mice. In fact, despite the resistant C57BL/6 background, Wdr73 CKO animals with only a single dose of ADR manifested increased albuminuria levels, GBM thickening, and podocyte effacement, all suggesting podocyte injury. Taken together, these findings strongly indicate that WDR73 plays key physiologic and cell biologic roles in focal adhesion.
PIP4K2C is ubiquitously expressed at different levels in tissues, with primary expression in the kidney, brain, heart, and testes [
15]. However, the role of PIP4K2C in kidney development and disease remains unclear. PIP4K2C can heterodimerize with PIP4K2A and has been suggested to serve as a chaperone for the more active isoforms, and its presence likely affects the distribution and enzyme activity of the 2A and 2B isoforms [
16,
27]. We confirmed that WDR73 can directly interact with PIP4K2C, and we found that the mRNA expression of PIP4K2C was higher in the mouse kidney than in other organs. WDR73 KO led to a significant reduction in the PIP4K2C and PIP4K2A proteins, resulting in the reduction in intracellular PIP2 and disruption of FA and actin filament assembly. These results may provide new clues regarding the role of interaction between PIP4K2C and WDR73 in the kidney.
Considering the reduction in PIP4K2C induced by WDR73 depletion, the stability of PIP4K2C was assessed. Our results revealed the unstable PIP4K2C should be eliminated mainly through enhanced autophagy, but not the ubiquitin–proteasome system. An autophagy protein EPG5 was subsequently proved to also interact with WDR73. EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes and is a key autophagy regulatory protein [
28]. We intended to further confirm these molecular interactions with WDR73 in vivo using mice for further verification.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






